Clear Sky Science · sv
rhinotypeR möjliggör reproducerbar tilldelning av rhinovirusgenotyper från VP4/2-sekvenser
Varför små förkylningsvirus fortfarande spelar roll
De flesta av oss ser på förkylningen som ett besvär snarare än ett allvarligt hot. Men de virus som orsakar många förkylningar — humana rhinovirus — är också kopplade till svåra lunginfektioner, astmaanfall och försämringar av kronisk lungsjukdom. För att följa hur dessa virus utvecklas och sprids behöver forskare sortera dem i precisa genetiska "typer", ungefär som att sätta streckkoder på produkter. Denna artikel presenterar rhinotypeR, ett fritt, öppen källkodsprogram som gör denna genetiska märkning mer exakt, konsekvent och lätt att upprepa, vilket hjälper folkhälsoteam att få bättre överblick över en ofta förbisedda familj av luftvägsvirus.
Den dolda mångfalden i vanliga förkylningar
Humana rhinovirus är extraordinärt vanliga och förekommer i upp till 60 % av prover från personer med plötslig luftvägssjukdom. De är långt ifrån ett enda virus — de delas in i tre huvudgrupper, kallade A, B och C, och minst 169 erkända genetiska typer. Olika typer beter sig olika: vissa kopplas oftare till svåra infektioner hos barn och till astmaförsämringar, medan andra ses mer sällan vid allvarlig sjukdom. Eftersom dessa typer utvecklas oberoende och bär distinkta ytegenskaper behöver forskare tillförlitliga metoder för att skilja dem åt om de vill följa hur utbrott rör sig genom skolor, hushåll och samhällen.
Från splittrade verktyg till en tydlig väg
Hittills har det varit ett lapptäcke att tilldela en rhinovirustyp utifrån dess genetiska kod. Forskare har vanligtvis fokuserat på en kort del av virusets genom som kallas VP4/2-regionen, jämfört den med kända referensstammar, mätt hur olika sekvenserna var och sedan tillämpat tröskelvärden för att avgöra vilken typ varje prov tillhörde. Men dessa steg gjordes med en blandning av olika program, manuella redigeringar och personlig bedömning. Det gjorde det svårt att jämföra eller upprepa olika studier, även när de använde liknande data. rhinotypeR skapades specifikt för att omvandla denna flerstegs-, felbenägna process till ett enda skriptat arbetsflöde som vem som helst kan köra och dela.
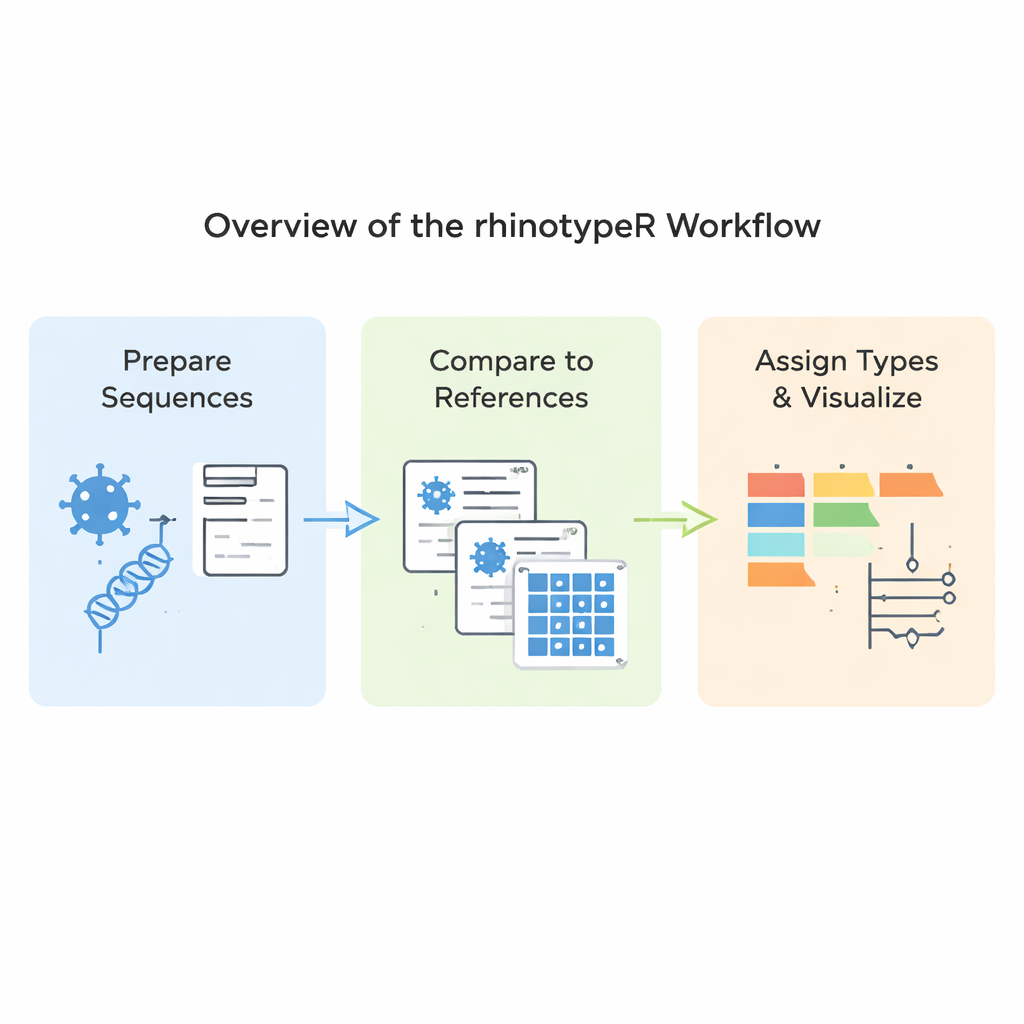
Vad den nya programvaran faktiskt gör
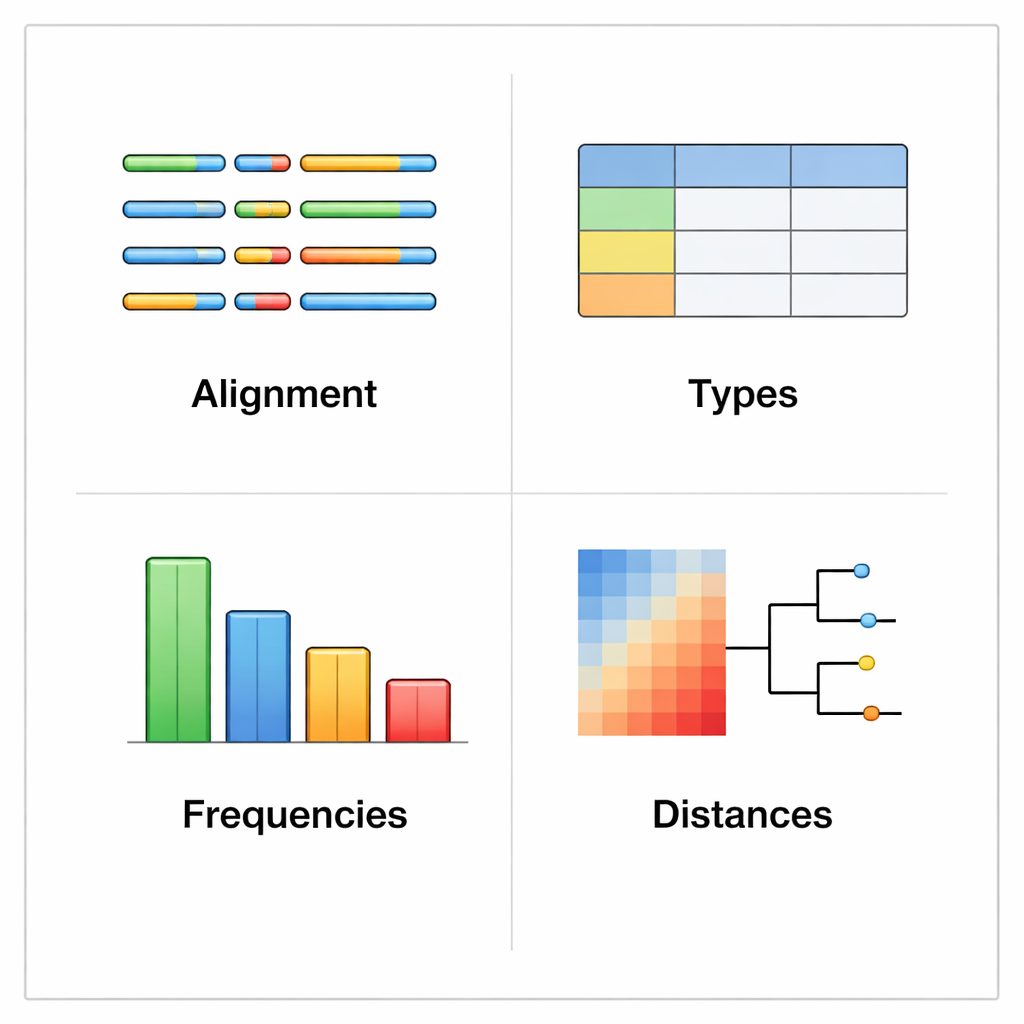
rhinotypeR körs i den välanvända R- och Bioconductor-miljön för dataanalys. Den tar in en samling rhinovirus VP4/2-sekvenser och leder dem genom tre huvudsteg: förberedelse och inpassning av sekvenserna, beräkning av hur långt var och en ligger från en kurerad uppsättning referenstyper, och sedan tilldelning av varje prov till närmaste kända typ eller markering som "otilldelad" om det är för olikt. Samma verktyg kan skapa visuella resultat, inklusive färgkodade kartor över genetiska skillnader, enkla fylogenetiska träd och diagram som visar hur vanliga olika typer är i en datamängd. Användare kan välja att utföra inpassning med externa program om de föredrar det, eller låta rhinotypeR hantera hela processen inom R för maximal reproducerbarhet.
Sätta verktyget på prov
För att kontrollera att rhinotypeR ger tillförlitliga resultat jämförde författarna dess avståndsberäkningar med dem från två etablerade program, ape och MEGA X, med samma indatafiler och modeller. Resultaten överensstämde nästan perfekt; eventuella små skillnader berodde på normal avrundning i datorberäkningar, inte faktiska metodskiljaktigheter. Teamet körde sedan rhinotypeR på en stor samling på mer än 2 300 rhinvirussekvenser från flera tidigare studier, och täckte över 90 % av kända typer. I ungefär fyra av fem fall överensstämde det nya verktyget exakt med tidigare typbeteckningar. De flesta avvikelserna låg precis runt de förbestämda tröskelvärden som används för att skilja en typ från en annan, vilket är precis där gränsfall förväntas. Viktigt är att prover som inte kunde tilldelas en känd typ med säkerhet inte uppvisade tecken på att bara vara lågkvalitativa eller ha låg virusmängd, vilket tyder på att de kan återspegla verklig viral mångfald.
Varför detta är viktigt för folkhälsan
För icke-specialister är huvudbudskapet att rhinotypeR inte uppfinner om hur forskare klassificerar förkylningsvirus; istället gör det processen tydligare, mer transparent och enklare att upprepa. Genom att paketera inpassning, avståndsberäkningar och typbestämning i ett öppet paket — tillsammans med tydliga visuella sammanfattningar — hjälper det forskare och övervakningsprogram att bearbeta tusentals prover på ett konsekvent sätt. Denna konsekvens förbättrar vår förmåga att jämföra studier från olika platser och tider, upptäcka ovanliga eller framväxande viruslinjer tidigt och koppla genetiska mönster till verkliga sjukdomstrender. På lång sikt stärker verktyg som rhinotypeR rutinövervakningen av till synes vanliga förkylningar som hos många kan utlösa allvarlig sjukdom.
Citering: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Nyckelord: rhinovirusgenotypning, molekylär övervakning, VP4/2-sekvensering, bioinformatiska verktyg, respiratoriska virus