Clear Sky Science · sv
En maskininlärningsmetod för att förutsäga osmotiska koefficienter och härleda aktivitetskoefficienter i alkylammoniumsalter
Vardagliga kemikalier med dold komplexitet
Från mjukmedel och hårbalsam till desinfektionsservetter och munskölj—en familj av kemikalier som kallas kvartära ammoniumsalter, ofta förkortade till ”Quats”, driver tyst många produkter vi förlitar oss på. De hjälper till att döda mikroorganismer, göra kläder mjukare och påskynda industriella reaktioner. Ändå har det visat sig svårt att förutsäga exakt hur dessa salter beter sig i vatten, vilket begränsar hur effektivt vi kan utforma säkrare och mer miljövänliga formuleringar. Denna studie visar hur modern maskininlärning kan lära sig från tidigare mätningar för att förutsäga det beteendet mer flexibelt och i många fall mer noggrant än traditionella modeller.
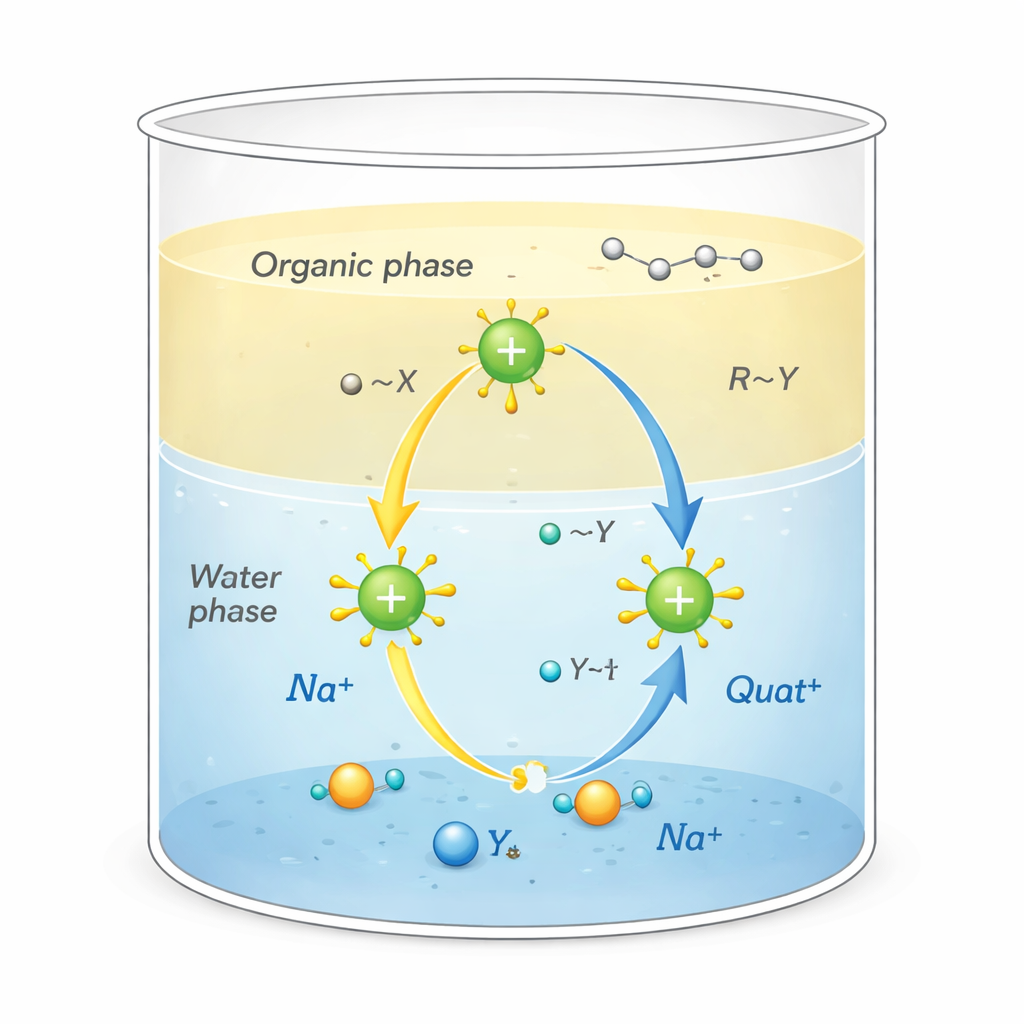
Varför dessa salter är viktiga
Quats är positivt laddade molekyler omgivna av kolrika ”svansar”. Denna ovanliga form gör att de kan utföra flera uppgifter samtidigt: fastna i fet smuts, fästa vid ytor som tyg eller hår och störa mikrobers membran, vilket gör dem till kraftfulla desinfektionsmedel och tensider. De används också som fasöverföringskatalysatorer, i praktiken som transportörer som för reaktiva joner från vatten in i oljeliknande lösningsmedel där de normalt inte skulle gå. Denna transport vid gränsytan mellan vatten och olja kan dramatiskt öka hastigheten för kemiska reaktioner som används vid tillverkning av läkemedel, polymerer och finare kemikalier.
Varför det är svårt att förutsäga deras beteende
För att kunna konstruera nya Quats eller justera befintliga behöver kemister veta hur de beter sig i lösning—hur starkt de interagerar med vatten och med andra lösta joner. Två nyckelmått är den osmotiska koefficienten, som speglar hur salter påverkar vattnets benägenhet att dras över membran, och aktivitetskoefficienten, som fångar hur ”effektiv” en löst art är jämfört med en ideal, perfekt blandad lösning. Traditionellt erhålls dessa värden antingen genom tidskrävande experiment eller genom komplexa fysikaliska modeller som Elektrolyt‑NRTL och Utökad UNIQUAC, vilka kräver många inpassade parametrar och inte enkelt kan generaliseras till nya molekyler.
Att lära en dator att läsa molekyler
Forskarna valde en annan väg: de undrade om en dator kunde lära sig sambandet mellan Quat‑struktur och osmotiskt beteende direkt från befintliga data. De samlade 1 654 mätningar av osmotiska koefficienter för 52 olika Quats från vetenskaplig litteratur. Varje molekyl beskrevs med SMILES‑notation—en strängrepresentation som kodar egenskaper såsom antal kol‑ och syreatomer, närvaro av bensenringar, förgreningar och typen av positivt laddad kvävegrupp, tillsammans med motsvarande negativ jon (som klorid, bromid eller nitrat). Dessa strukturella deskriptorer, plus salthalten, fungerade som indata till flera övervakade maskininlärningsalgoritmer implementerade i Python.
Att hitta den mest pålitliga prediktorn
Sju olika algoritmer—inklusive linjär regression, beslutsträd, random forests, supportvektormaskiner, gradient boosting, k‑närmsta grannar och Gaussiska processer—tränades på 70% av datan och testades på de återstående 30%. Teamet använde också ett striktare valideringsschema där all data för ett salt hölls ut för att se hur väl modellerna exrapolerade till en verkligt otestad förening. Linjär regression presterade dåligt och missade viktiga icke‑linjära trender. Trädbaserade metoder anpassade sig mycket väl till träningsdatan men gav något ojämna förutsägelser och tappade i noggrannhet för nya salter. Den Gaussiska processmodellen hittade den bästa balansen: den levererade släta, fysiskt rimliga kurvor för osmotiska koefficienter och uppnådde ett genomsnittligt absolut procentfel på omkring 5% totalt, och överträffade alternativa maskininlärningsmetoder under de tuffaste testerna.
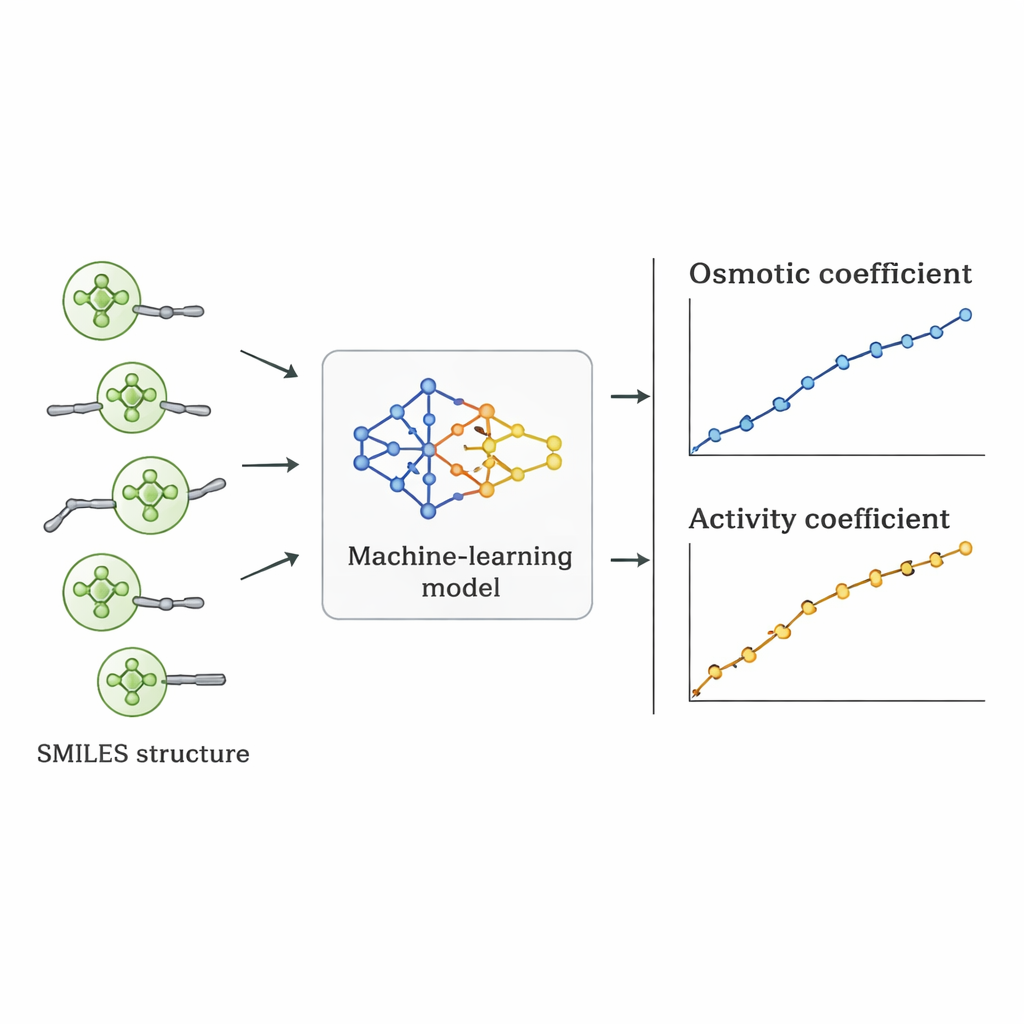
Från osmotiskt beteende till användbara designtal
När den bästa modellen valts omvandlades dess förutsagda osmotiska koefficienter till aktivitetskoefficienter med hjälp av standardtermodynamiska samband. När dessa aktivitetskoefficienter jämfördes med värden härledda från experiment och etablerade fysikaliska modeller matchade eller överträffade maskininlärningsmetoden ofta dessa för enskilda Quats. Även om dess genomsnittliga fel över alla ämnen var något högre än hos vissa specialiserade modeller hade den en avgörande fördel: eftersom den drivs av strukturella deskriptorer snarare än salt‑specifik inpassning kan den tillämpas på nya Quats som aldrig mätts i labbet, så länge deras strukturer liknar dem i träningsdatasetet.
Vad detta betyder för produkter och processer
För en icke‑specialist är budskapet att datorer nu kan ”läsa” kompakta textbeskrivningar av molekyler och, utifrån mönster inlärda från tidigare data, förutsäga hur dessa molekyler beter sig i vatten med imponerande noggrannhet. Detta öppnar dörren för snabbare, billigare screening av nya Quats för desinfektionsmedel, rengöringsmedel, personvårdsprodukter och industriella katalysatorer, utan att varje kandidat kräver uttömmande experiment. Den nuvarande modellen är bara ett första steg, och författarna noterar att rikare molekylära fingeravtryck och nyare algoritmer skulle kunna förbättra prestandan ytterligare. Fortfarande visar den hur datadrivna verktyg kan komplettera traditionell kemi och hjälpa ingenjörer att utforma mer effektiva och potentiellt säkrare formuleringar genom att utforska kemiska möjligheter som vore opraktiska att testa en och en i laboratoriet.
Citering: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Nyckelord: kvartära ammoniumsalter, fasöverföringskatalys, osmotiska koefficienter, aktivitetskoefficienter, maskininlärning i kemi