Clear Sky Science · sv
ATF4 reglerar mitokondriell dysfunktion och mitofagi, vilket bidrar till apoptos i korneal endotel
Varför ögats fönster kan bli grumligt
Våra hornhinnor — ögats klara främre fönster — förblir genomskinliga tack vare ett tunt, hårt arbetande cellager på deras inre yta. Vid Fuchs endotelial korneal dystrofi (FECD) förlorar miljontals människor gradvis dessa celler, vilket leder till svullnad, grumlig syn och ofta hornhinnetransplantationer. Denna studie ställer en grundläggande men avgörande fråga: vad får dessa celler att börja dö, och skulle avstängning av en molekylär ”omkopplare” kunna rädda dem?
Ett känsligt cellager som håller synen klar
Korneaendotelet är ett enda skikt av sexkantiga celler som ständigt pumpar ut vätska ur hornhinnan för att hålla den klar. Vid FECD blir dessa celler stressade och försvinner gradvis, samtidigt som knölar av onormalt material, kallade guttae, byggs upp på den underliggande membranen. Eftersom det inte finns några godkända läkemedel för FECD och hornhinnetransplantationer är huvudbehandlingen försöker forskare förstå exakt hur stress inne i dessa celler driver dem mot död. Tidigare arbete pekade var för sig på påfrestningar i två nyckelkompartiment — endoplasmatiska nätverket (cellens proteinvikningsfabrik) och mitokondrierna (cellens kraftverk) — men hur dessa två stressvarningssystem kommunicerar med varandra var fortfarande oklart.
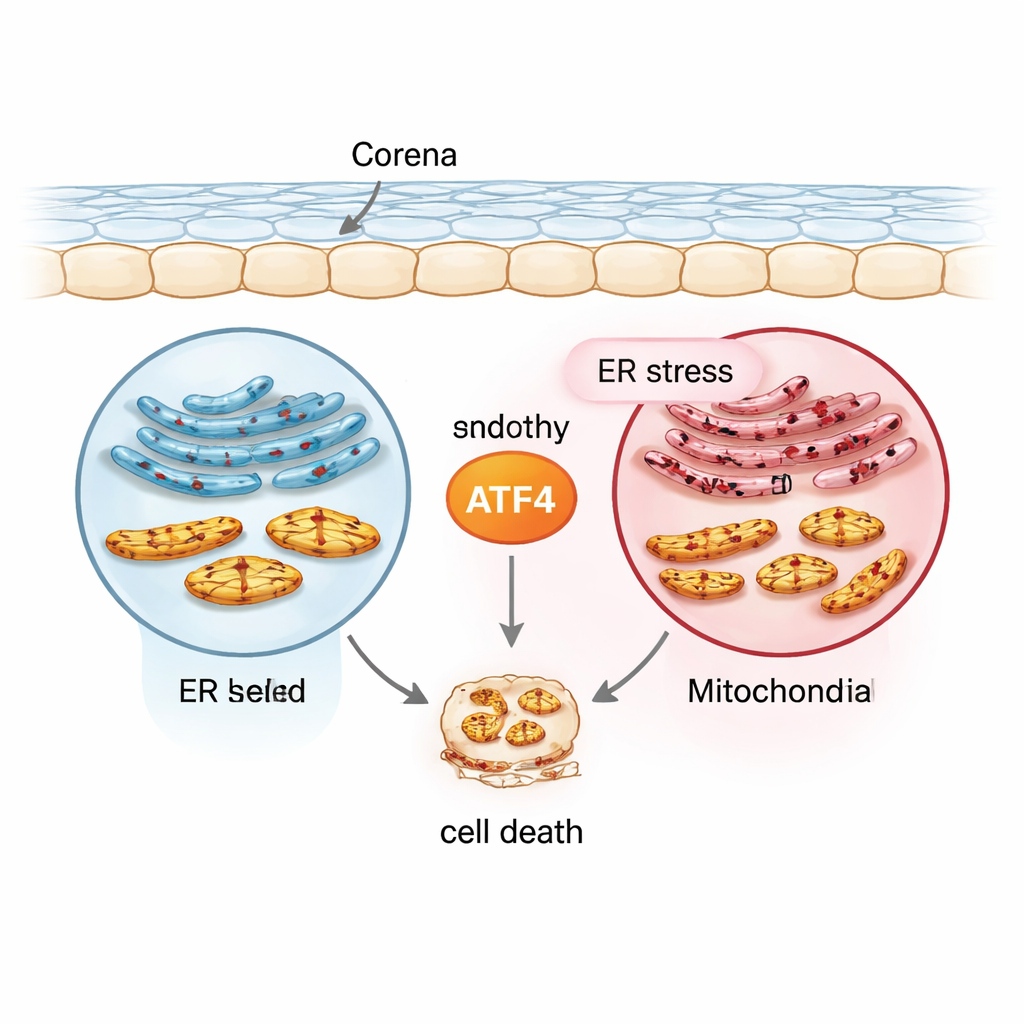
Stressbudbäraren i centrum: ATF4
Teamet fokuserade på ett protein kallat ATF4, en transkriptionsfaktor som slår på eller av många stressresponsgener. Med en normal human korneal endotelcellinje (21T), en FECD‑lik cellinje som bär den sjukdomsrelaterade TCF4‑repeatexpansionen (F35T), primära humana korneaendotelceller och musemodeller utsatta för ultraviolett A (UVA)‑ljus skapade de en rad förhållanden som efterliknar kronisk stress. De utlöste endoplasmatiskt retikelstress med ett läkemedel kallat tunicamycin och mätte sedan ATF4 och andra markörer. Jämfört med normala celler hade FECD‑lika celler redan från början högre nivåer av ATF4 och relaterade stressproteiner, och ATF4 ökade ännu mer under kronisk stress både i odlade celler och i humana korneala vävnader. Detta mönster placerade ATF4 i korsningen mellan tidiga skyddande svar och senare, självdestruktiv signalering.
Från strömavbrott till programmerad död
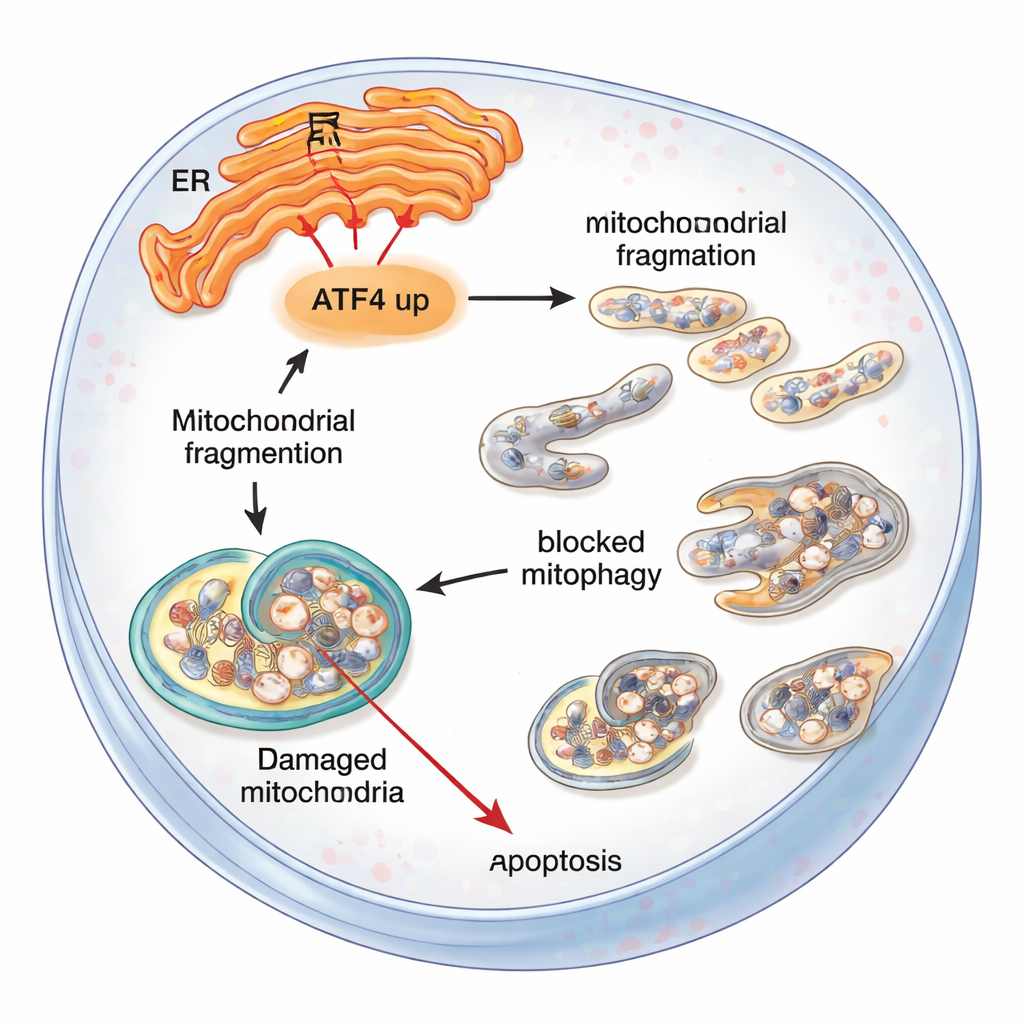
Därefter undersökte forskarna hur denna stress påverkade mitokondrierna. I FECD‑lika celler producerade mitokondrierna mindre ATP, förlorade sin elektriska membranpotential och sprack upp från långa, nätverksliknande former till många små fragment. Dessa förändringar förvärrades när endoplasmatiskt retikelstress var långvarig. Samtidigt blev klassiska celldödsproteiner — såsom aktiverade kaspaser och DNA‑reparationsproteinet PARP i sin klyvda, pro‑dödsform — mer förekommande, medan skyddande proteiner som Bcl‑2 minskade. Tillsammans tyder dessa förändringar på att stressade korneaendotelceller i FECD drivs mot mitokondrie‑driven apoptos, en prydlig men irreversibel form av programmerat celldöd.
Rengöringssystemet fastnar vid kronisk stress
Under normala förhållanden avlägsnas kraftigt skadade mitokondrier genom en återvinningsprocess kallad mitofagi, där de märks och omsluts i små blåsor för borttransport. Teamet fann att tidiga mitofagi‑"startmolekyler" (Parkin och LC3) aktiverades i både normala och FECD‑lika celler, särskilt efter stress. Men viktiga stödjande proteiner var minskade, och elektronmikroskopi visade en uppbyggnad av delvis nedbrutna mitokondrier fångade i vesiklar. Detta antyder att även om rengöringsprocessen startade, misslyckades den att slutföras, vilket lämnade cellerna fulla av defekta kraftverk som ytterligare driver på stress och död istället för återhämtning.
Stänga av ATF4 för att rädda celler
För att testa om ATF4 drev denna spiral använde forskarna små interfererande RNA för att partiellt tysta ATF4 i odlade korneaendotelceller. Under samma kroniska stress visade celler med reducerat ATF4 lägre nivåer av dödsfrämjande proteiner, friskare mitokondriell membranpotential, mindre fragmentering och bättre överlevnad i viabilitetstester. Viktigt var att antalet fastnade mitofagistrukturer minskade, vilket tyder på att sänkning av ATF4 hjälpte till att återställa en mer effektiv balans mellan skada och städning. I möss konstruerade för att bara ha en fungerande kopia av ATF4‑genen orsakade UVA‑exponering mindre aktivering av en pro‑döds‑partnerprotein, CHOP, och bevarade fler normalt formade endotelceller jämfört med möss med full ATF4‑funktion.
Vad detta betyder för personer med FECD
För icke‑specialister är budskapet att en stressbudbärare, ATF4, kan väga över korneaendotelceller från att klara av stress till att kollapsa. När endoplasmatiskt retikelstress är långvarig bidrar ATF4 till att störa mitokondrierna, få städmaskineriet att fastna och uppmuntrar till sist dessa viktiga celler att begå självmord. Att dämpa ATF4 — antingen genetiskt i möss eller med riktade molekylära verktyg i celler — skyddar mitokondrier, förbättrar avfallshanteringen och håller fler celler vid liv. Även om detta arbete fortfarande befinner sig på laboratorie‑ och djurstadiet framhäver det ATF4 och relaterade stressvägar som lovande läkemedelsmål som en dag skulle kunna bromsa eller förhindra utvecklingen av Fuchs dystrofi och minska behovet av hornhinnetransplantationer.
Citering: Qureshi, S., Kim, S.Y., Lee, S. et al. ATF4 regulates mitochondrial dysfunction and mitophagy, contributing to corneal endothelial apoptosis. Sci Rep 16, 5960 (2026). https://doi.org/10.1038/s41598-026-36453-x
Nyckelord: Fuchs endotelial korneal dystrofi, korneal endotel, mitokondriell stress, mitofagi, ATF4