Clear Sky Science · sv
Hybridt djupinlärningsramverk för noggrann klassificering av högdimensionella genomiska data
Att förstå floden av genomdata
Moderna DNA-tekniker kan mäta tiotusentals gener i ett enda experiment, vilket lovar tidigare sjukdomsdetektion och mer precisa behandlingar. Ändå är denna datarika miljö så stor, brusig och komplex att även kraftfulla datormodeller ofta har svårt att hitta klara, pålitliga mönster. Denna artikel presenterar en ny typ av artificiell intelligens (AI) som är särskilt utformad för att hantera sådana överväldigande genomiska data, med målet att göra prediktioner mer träffsäkra samtidigt som den förklarar hur dessa prediktioner uppstod.
Varför genomiska data är svåra att använda
Genomiska studier producerar rutinmässigt långt fler mätningar än det finns patienter eller prover. Många av dessa mätningar är irrelevanta, redundanta eller förvrängda av tekniskt brus. Traditionella maskininlärningsmetoder kräver antingen att mänskliga experter manuellt väljer vilka gener som kan vara viktiga, eller så försöker de använda allt och riskerar överanpassning — det vill säga att prestera bra på träningsdata men misslyckas på nya fall. Djupinlärning, som har revolutionerat områden som bildigenkänning, kan automatiskt lära sig mönster från rådata. Men inom genomik beter den sig ofta som en svart låda: den kan ge korrekta svar men erbjuder lite insikt i varför, vilket begränsar dess acceptans inom medicin där transparens är avgörande.
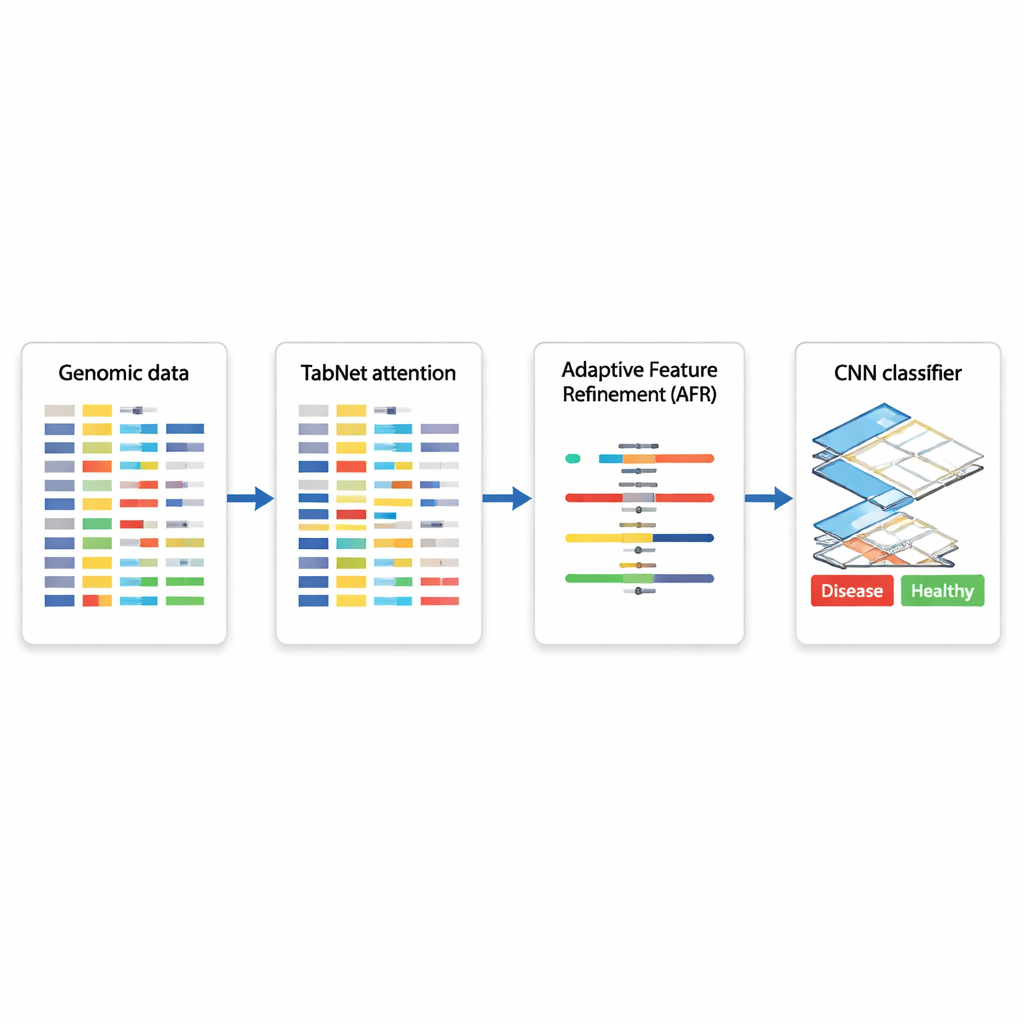
En hybrid-AI-arkitektur för genbaserade beslut
Författarna föreslår en hybrid djupinlärningsarkitektur som kedjar ihop tre specialiserade moduler. Först fungerar en komponent kallad TabNet som en strålkastare, skannar alla tillgängliga genomiska mätningar och lär sig vilka funktioner som är mest informativa för en given uppgift — till exempel att skilja cancerösa från icke-cancerösa vävnader. Istället för att behandla varje gen lika fokuserar TabNet uppmärksamheten på en gles delmängd som verkar mest relevant. Därefter tar ett lager för Adaptiv Funktionsförfining (AFR) dessa utvalda signaler och omviktar dem, förstärker konsekventa, meningsfulla mönster samtidigt som brus ytterligare dämpas. Slutligen undersöker ett konvolutionellt neuralt nätverk (CNN), vanligt inom bildanalys, hur de förfinade funktionerna interagerar lokalt och fångar subtila relationer mellan grupper av gener som kan indikera en viss sjukdomssubtyp eller biologiskt tillstånd.
Att testa modellen
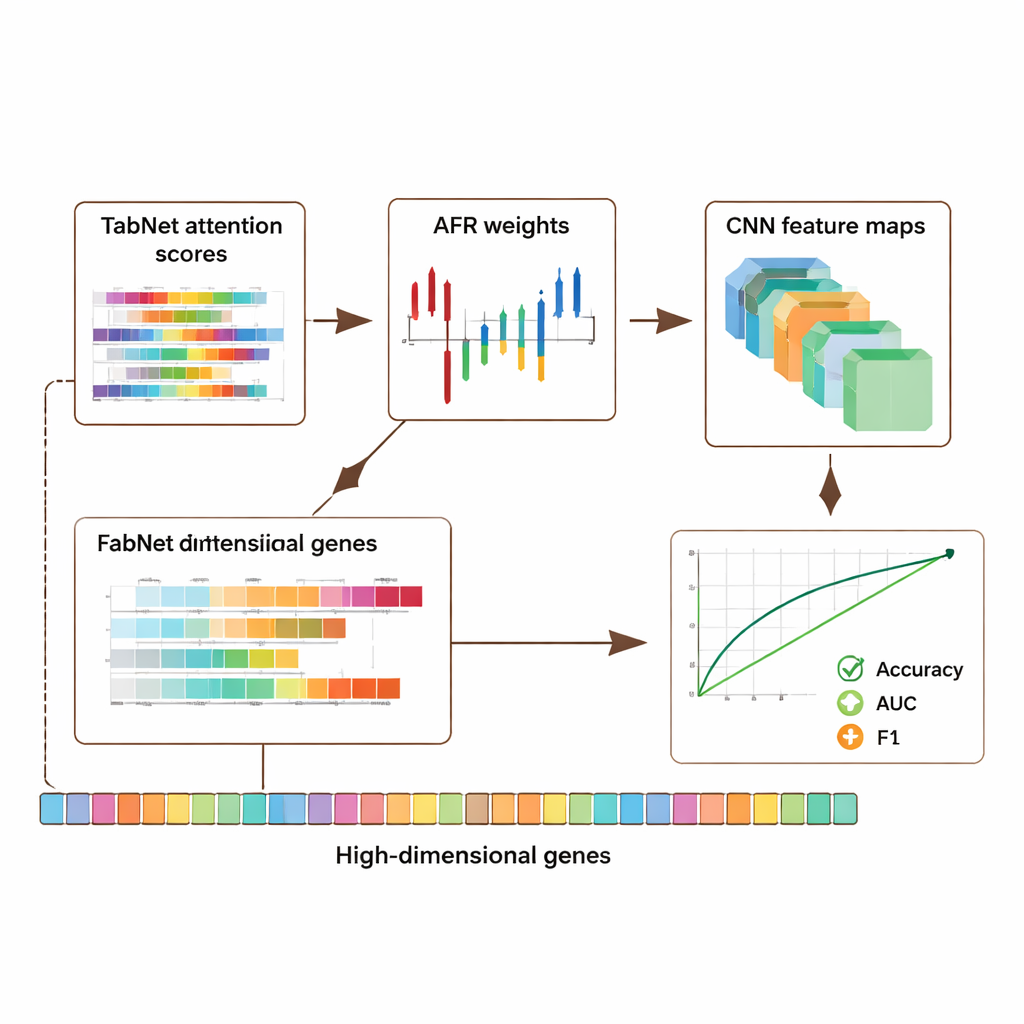
Ramverket utvärderades på tre stora offentliga resurser: en bröstcancersats från The Cancer Genome Atlas, ett single-cell-melanomdataset från Gene Expression Omnibus och ett epigenomiskt dataset från ENCODE-projektet. Tillsammans omfattar dessa samlingar tusentals prover och tiotusentals features per prov, vilka täcker genaktivitet och kemiska markörer på DNA. Över alla dataset överträffade den hybrida modellen flera toppmoderna metoder och förbättrade noggrannhet och nyckelmått för klassificering som area under ROC-kurvan (AUC) och F1-poäng med ungefär 5–8 procentenheter. Viktigt är att dessa förbättringar inte skedde på bekostnad av transparens: modellen producerar uppmärksamhetskartor från TabNet och aktiveringskartor från CNN som framhäver vilka gener och regioner som varit mest inflytelserika i varje prediktion.
Att balansera noggrannhet, sekretess och förtroende
Där genomiska data är djupt personliga studerade författarna även hur man skyddar sekretessen samtidigt som man behåller användbar signal. De introducerade en adaptiv sekretessmekanism som adderar mer brus till högkänsliga funktioner och mindre till andra, kombinerat med maskering av utvalda ingångar. Tester visade att även när måttligt brus infördes bibehöll modellen stark noggrannhet och diskriminering, med en gradvis försämring av prestanda i takt med att skyddet skärptes. Samtidigt pekade de tolkbara uppmärksamhets- och aktiveringsmönstren ofta mot gener som redan är kända för att spela roller i cancer och immunsreglering, vilket tyder på att systemet inte enbart memorerar data utan fångar biologiskt meningsfulla signaler. En ablationsstudie — där delar av arkitekturen systematiskt togs bort — bekräftade att varje modul, särskilt AFR-lagret, gjorde en mätbar insats för prestandan.
Vad detta innebär för framtidens medicin
Enkelt uttryckt erbjuder detta arbete ett smartare sätt att sålla igenom enorma genomiska kalkylblad för att hitta mönster kopplade till sjukdom, samtidigt som det visar vilka poster i kalkylbladet som spelade störst roll. Genom att kombinera riktad funktionsselektion, noggrann förfining och mönsterigenkänning förbättrar den hybrida modellen prediktionsnoggrannheten, förblir hanterbar beräkningsmässigt och tillhandahåller visuella ledtrådar som kliniker och biologer kan tolka. Även om mer testning krävs på bredare och mer mångsidiga patientgrupper, kan sådana ramverk hjälpa till att identifiera nya biomarkörer, förfina sjukdomssubtyper och stödja kliniska beslutsverktyg inom precision medicine — vilket för AI-baserad DNA-analys ett steg närmare verklig användning.
Citering: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Nyckelord: genomisk djupinlärning, upptäckt av cancerbiomarkörer, tolkningsbar AI, precision medicine, sekretessbevarande genomik