Clear Sky Science · sv
En kvantitativ studie av cytotoxiska föreningar med grafbaserade deskriptorer och maskininlärning
Varför denna forskning är viktig för framtida cancerläkemedel
Cancerläkemedel som dödar tumörceller, så kallade cytotoxiska läkemedel, balanserar ofta mellan att rädda liv och att orsaka allvarliga biverkningar. För att utforma säkrare och mer effektiva behandlingar behöver forskare snabba, tillförlitliga sätt att förutsäga hur dessa läkemedel rör sig i kroppen — hur väl de tas upp, hur lätt de korsar cellmembran och var de hamnar. Denna studie visar hur matematiska beskrivningar av läkemedelsmolekyler, kombinerade med modern maskininlärning, kan uppskatta en nyckelstorhet som styr detta beteende med hög noggrannhet, vilket potentiellt kan påskynda sökandet efter bättre cancerterapier.
En viktig yta som styr vart läkemedel kan ta vägen
En central idé i artikeln är den topologiska polära ytan, eller Top_PSA. Enkelt uttryckt är detta ett tal som speglar hur stor del av en molekyls yta som består av ”polära” regioner — delar som gillar vatten och kan bilda vätebindningar. Molekyler med mycket hög polar yta har ofta svårt att passera fettiga cellmembran och kan vara dåligt absorberade vid oral administrering. Molekyler med mycket låg polar yta kan å andra sidan tränga igenom barriärer för lätt, vilket ibland ger oönskade effekter i känsliga vävnader som hjärnan. Top_PSA har blivit en populär genväg för att uppskatta dessa transportegenskaper eftersom den snabbt kan beräknas från en 2D-ritning av en molekyl utan att behöva långsamma 3D-simuleringar.
Att omvandla molekylritningar till siffror
Forskarna sammanställde en kurerad uppsättning på 156 olika cytotoxiska föreningar hämtade från verkliga anticancerläkemedel och experimentella ämnen. De omvandlade sedan varje molekyl till 58 så kallade deskriptorer — tal som fångar egenskaper såsom antal atomer, antal ringar, hur flexibla bindningarna är, hur många atomer som kan bilda vätebindningar och hur polära eller elektronegativa olika delar är. Många av dessa deskriptorer kommer från grafteori, där en molekyl behandlas som ett nätverk av sammankopplade noder och länkar. Denna rika numeriska porträttering av varje molekyl tjänade som indata för datorbaserade modeller som försökte förutsäga Top_PSA-värden beräknade med välanvända kemiverktyg.
Att testa flera vägar till träffsäker förutsägelse
För att hitta det bästa sättet att koppla dessa deskriptorer till Top_PSA jämförde teamet flera modelleringsstrategier. De provade standard linjär regression samt två ”regulariserade” varianter kallade ridge och LASSO-regression, som är utformade för att bättre hantera bullrig, överlappande information. De utforskade också olika datapreparationsscheman: att passa modeller direkt på råa deskriptorer, komprimera dem med principal component analysis (PCA), skala dem på ett sätt som minskar effekten av extrema värden (robust skalning), justera uteliggare och beskära starkt korrelerade funktioner med hjälp av en måttstock kallad variance inflation factor. Varje angreppssätt utvärderades noggrant med k-fold cross-validation, en metod som upprepat delar upp data i tränings- och testdelar för att skydda mot överanpassning.
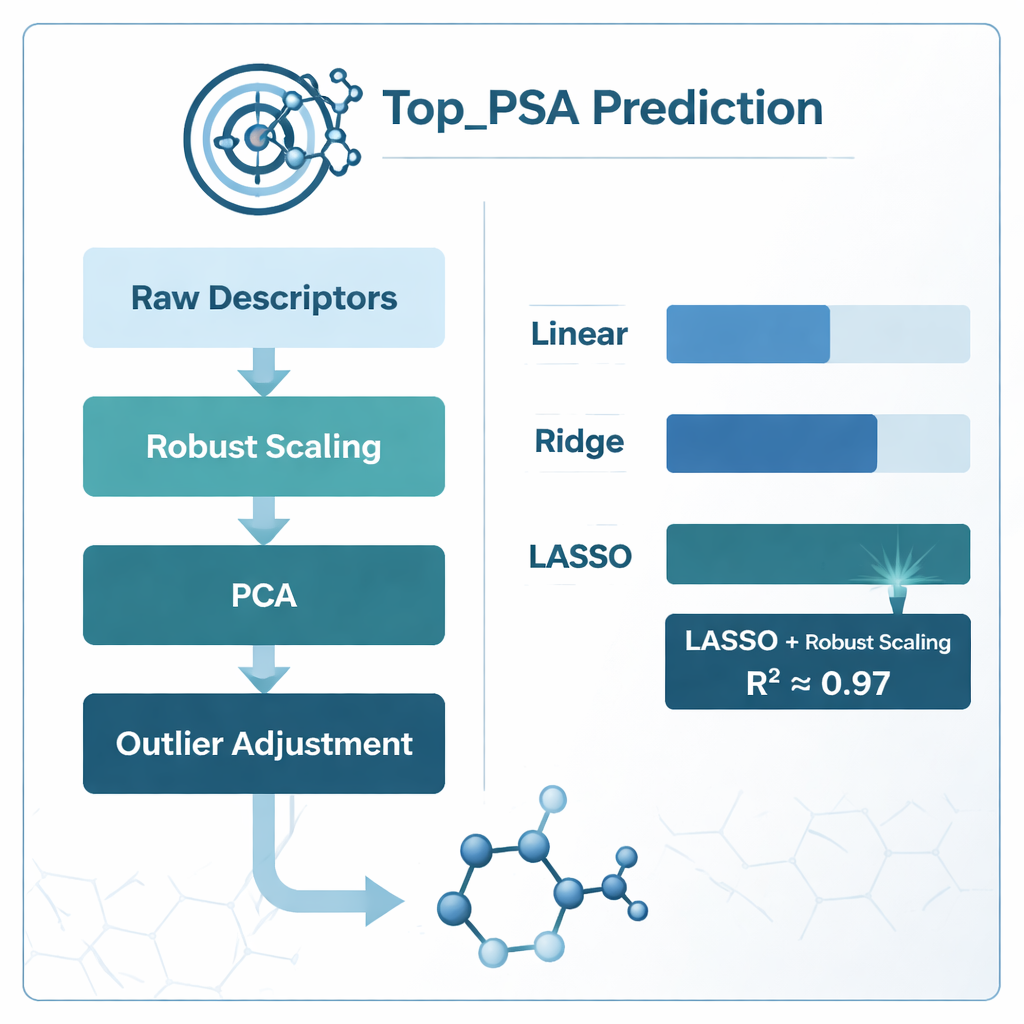
Vad som fungerade bäst och vad modellerna lärde sig
Den tydliga vinnaren var kombinationen av robust skalning och LASSO-regression, som uppnådde en förklaringsgrad (R²) på cirka 0,97 — vilket betyder att den kunde förklara ungefär 97 % av variationen i Top_PSA över de 156 läkemedlen. PCA-baserade modeller kom nära i rå noggrannhet men var svårare att tolka kemiskt eftersom de ursprungliga deskriptorerna blandas samman till abstrakta komponenter. Enkel beskärning av korrelerade deskriptorer med variance inflation factor försämrade faktiskt prestandan, vilket tyder på att vissa överlappande mått ändå bär användbar kemisk information. Genom att undersöka vilka deskriptorvikter LASSO behöll som icke-noll fann författarna att de viktigaste faktorerna var förekomsten av heteroatomer som kväve och syre, förmågan att ge eller ta emot vätebindningar samt index som följer hur elektronegativa atomer är ordnade över molekylgrafen — alla egenskaper som stämmer överens med den intuitiva kemiska förståelsen av polar yta.
Hur detta kan vägleda bättre läkemedelsdesign
För läsare utanför fältet är huvudbudskapet att noggrant förberedda matematiska fingeravtryck av molekyler, när de paras med välvalda maskininlärningsmetoder, kan ge snabba och pålitliga uppskattningar av hur ”klibbiga” eller ”glidiga” cancerläkemedel kommer att vara när de färdas genom kroppen. Studien erbjuder praktisk vägledning för andra forskare om hur man förbehandlar deskriptordata, vilka modelleringsmetoder man bör föredra och vilka genvägar man bör undvika. På sikt kan sådana robusta, tolkbara modeller för Top_PSA hjälpa kemister att filtrera enorma virtuella bibliotek av potentiella läkemedel och rikta sina ansträngningar mot föreningar med rätt balans mellan membranpenetration och säkerhet — ett viktigt steg mot mer effektiva och mindre toxiska cancerbehandlingar.
Citering: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Nyckelord: cytotoxiska läkemedel, polar yta, molekylära deskriptorer, maskininlärning, läkemedelspenetration