Clear Sky Science · sv
Mitokondriell dysfunktion och Ca2+-dysreglering i mänskliga iPSC‑härledda neuroner med presenilin‑1‑mutation uppstår under stress via en MCU‑1‑oberoende mekanism
Varför detta är viktigt för Alzheimers sjukdom
Alzheimers sjukdom beskrivs ofta med hänvisning till klibbiga proteinkluster i hjärnan, men länge innan minnet sviktar kan de små ”kraftverken” inne i nervcellerna — mitokondrierna — och hanteringen av kalciumjoner redan börja gå fel. Denna studie använder mänskliga neuroner odlade från hudceller från en person som bär en välkänd familjär Alzheimer‑mutation för att ställa en enkel men avgörande fråga: hur tidigt, och på vilket sätt, börjar energiproduktionen och kalciumbalansen att svikta?
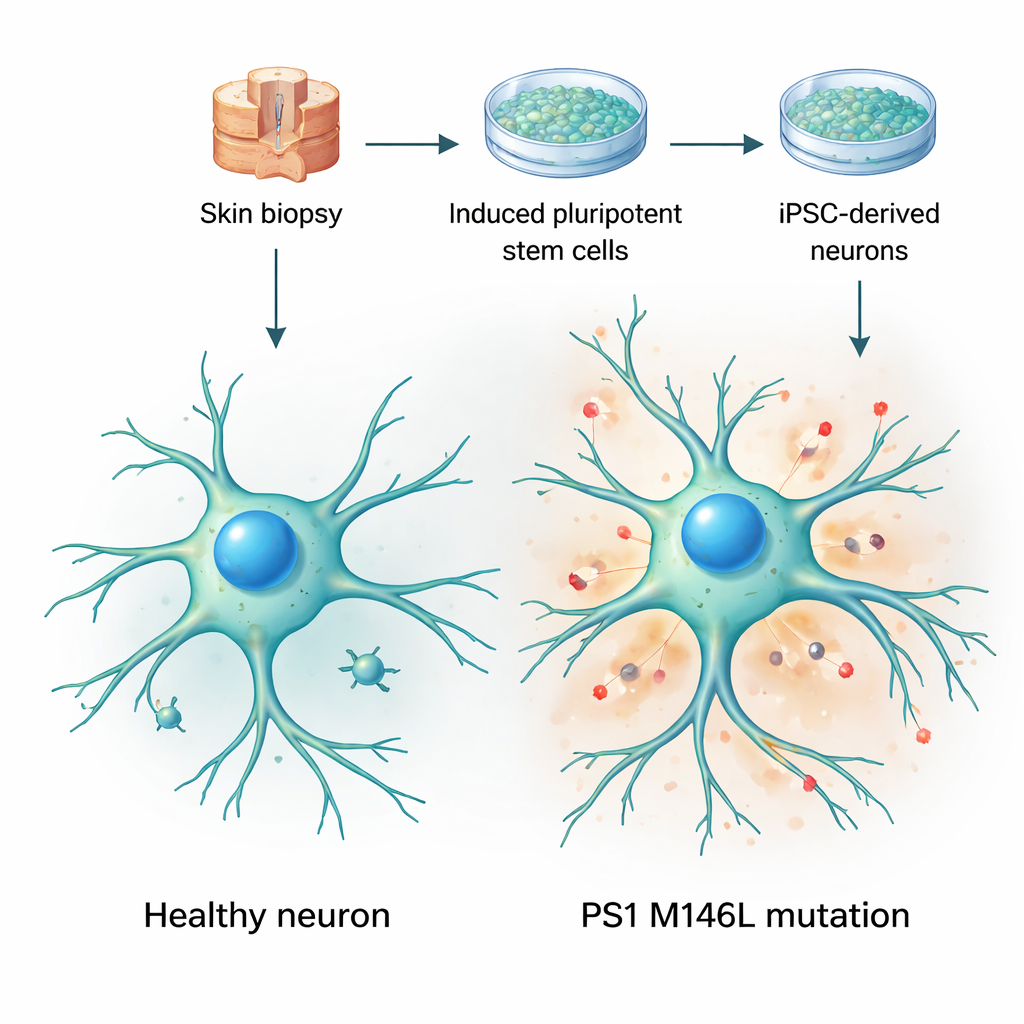
Att förvandla hudceller till levande hjärnmodeller
Forskarna började med hudbiopsier från två kvinnor: en frisk frivillig och en symtomfri bärare av en presenilin‑1‑mutation kallad M146L, som förekommer i en argentinsk familj med tidigt insjuknande i Alzheimer. De omprogrammerade hudcellerna till inducerade pluripotenta stamceller — celler som kan bli nästan vilken vävnad som helst — och styrde dem sedan att utvecklas till nervceller. Under flera veckor i odling antog dessa celler typiska neuronala former, förlängde långa, grenade utskott och uttryckte vanliga neuronala markörer. Viktigt är att både kontroll‑ och mutantcellerna mognade i liknande takt och såg i stort sett friska ut, vilket gjorde det möjligt för teamet att fokusera på subtila funktionella förändringar snarare än uppenbart cellbortfall eller skada.
Elektriska signaler och kalcium under påfrestning
Nervceller är beroende av strikt kontroll av kalcium, en laddad atom som fungerar som en snabb av‑ och på‑brytare för många cellulära processer. Med hjälp av fluorescerande färgämnen följde teamet hur kalciumnivåerna förändrades inne i cellerna när de elektriskt stimulerades med kalium eller aktiverades med signalsubstanser. Vid enkel depolariserande stimulans visade neuroner som bar M146L‑mutationen svagare kalciumökningar än kontrollneuroner, vilket tyder på problem med att upprätthålla de elektriska och joniska gradienter som normalt driver kalciuminflöde. Men när forskarna utlöste en mer stressande situation — genom att tvinga kalcium att läcka från interna lager i endoplasmatiska retiklet — blev skillnaden tydligare. Som svar på denna stress tog mitokondrier i mutanta neuroner upp märkbart mindre kalcium än de i kontrollceller, vilket indikerar en nedsatt förmåga att buffra farliga kalciumpikar.
Att koppla bort energianvändning från kalciumbalans
För att förstå hur denna förändrade kalciumhantering påverkar cellmetabolismen mätte forskarna hur mycket syre neuronerna konsumerade — en direkt proxy för mitokondriell aktivitet. Överraskande nog andades neuroner med M146L‑mutationen mer: deras basala och maximala syrgaskonsumtionshastigheter samt mängden syre kopplat till ATP‑produktion var alla högre än i kontrollceller. Ändå verkade effektiviteten i kopplingen mellan syreförbrukning och ATP‑produktion liknande, och det fanns ingen ökning i antalet mitokondrier eller i viktiga ATP‑producerande enzymer. Istället var mitokondrierna i mutanta neuroner längre och mer tubulära, med högre nivåer av ett fusionsprotein kallat mitofusin‑1, ett mönster som ofta ses i celler under kronisk, låggradig stress. Dessa hyperaktiva, förlängda mitokondrier genererade också mer reaktiva syreföreningar, instabila molekyler som kan skada proteiner och DNA om de inte kontrolleras ordentligt.
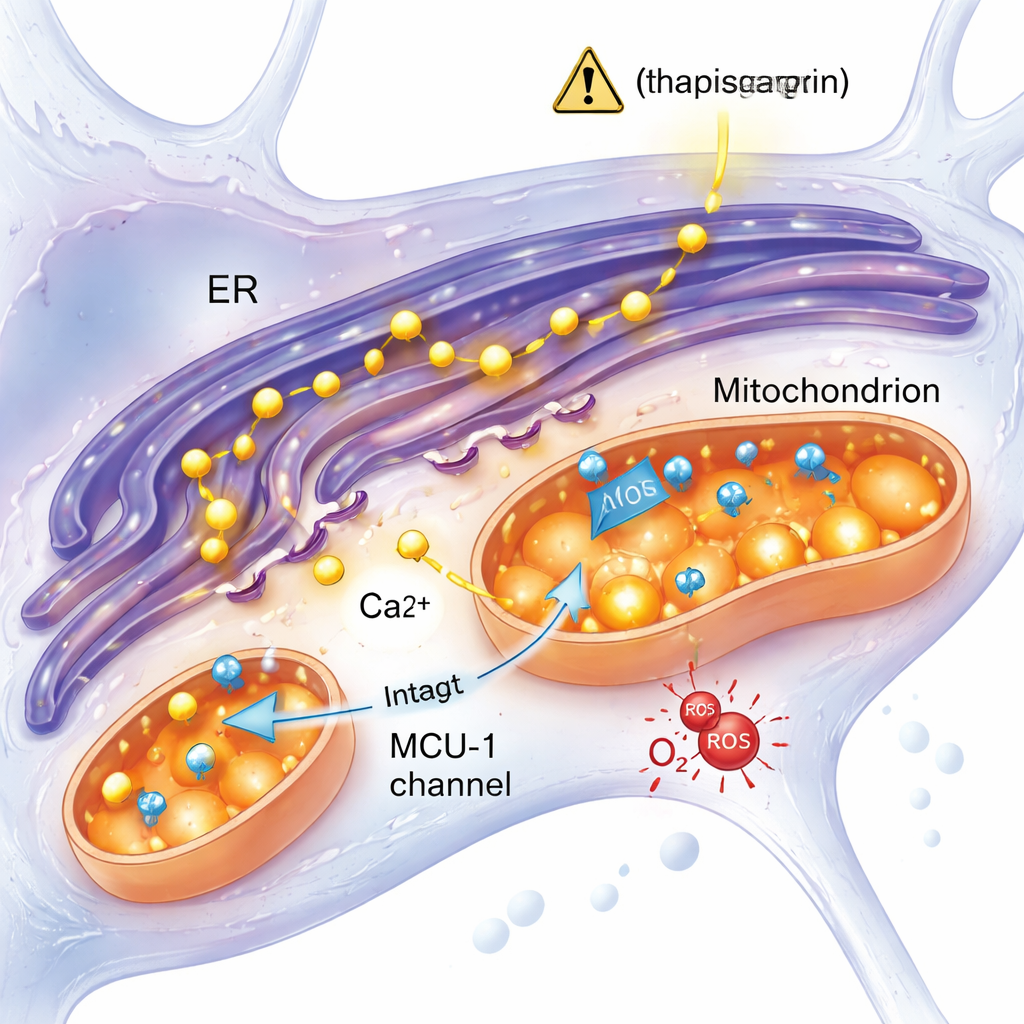
En stressreaktion oberoende av en nyckelkalciumkanal
En ledande idé inom Alzheimerforskning är att ett överskott av kalcium från endoplasmatiska retiklet forsar in i mitokondrierna genom en kanal kallad mitokondriell kalciumuniporter (MCU‑1), överbelastar dem och driver dysfunktion. Denna studie testade den föreställningen direkt. När teamet blockerade MCU‑1 med en specifik hämmare visade både kontroll‑ och mutantneuroner starka minskningar i mitokondriellt kalciumupptag, vilket bekräftar att kanalen i sig fungerade i båda grupperna. Dessutom, när kalciumfrigöring utlösts via en mer fysiologisk väg som involverar IP3‑receptorn — en annan viktig kalciumport — svarade mutant‑ och kontrollcellerna likartat. Dessa resultat talar emot en trasig MCU‑1‑kanal och tyder istället på att de fysiska och funktionella kontakterna mellan endoplasmatiska retiklet och mitokondrierna, eller andra aspekter av deras interaktion, är förändrade i de mutanta neuronerna.
Vad detta betyder för förståelse och behandling
Tillsammans målar fynden upp en bild av mänskliga neuroner som bär PS1 M146L‑mutationen som celler som ser normala ut i vila men reagerar onormalt under stress. Deras mitokondrier misslyckas med att ta upp tillräckligt med kalcium när interna lager plötsligt frigörs, samtidigt som de arbetar intensivare — konsumerar mer syre och genererar mer reaktiva syreföreningar — som om de fastnat i ett kostsamt kompensationsläge. Eftersom detta sker i levande mänskligt härledda neuroner innan några kliniska symtom visar sig, stöder arbetet idén att störd kalciumsignalering och tidig mitokondriell överbelastning är händelser uppströms i Alzheimers sjukdom, inte bara sena följdverkningar. För icke‑specialister är huvudbudskapet att bevarandet av balansen mellan kalciumsignaler och mitokondriell energiproduktion kan vara lika centralt för att förebygga sjukdom som att rikta in sig på de bättre kända amyloida placken.
Citering: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Nyckelord: Alzheimers sjukdom, mitokondrier, kalciumsignalering, presenilin‑1‑mutation, iPSC‑härledda neuroner