Clear Sky Science · sv
Minimalt virtuellt dataset för reproducerbar de novo-assemblans av triploida genom
Varför trekopiers-genom är viktiga
Många grödor och andra organismer bär inte bara två kopior av varje kromosom, som människor gör—de kan ha tre eller fler. Att pussla ihop dessa extra kopior från DNA-sekvenseringsdata är förvånansvärt svårt, eftersom kopiorna är mycket lika men inte helt identiska. Denna artikel introducerar ett litet men noggrant utformat ”virtuellt” dataset som gör det möjligt för forskare att testa och jämföra genomsamlingsprogram på ett realistiskt trekopiersproblem (triploid), under förhållanden som är helt kända och reproducerbara.
Att bygga ett enkelt ersättningsgenom
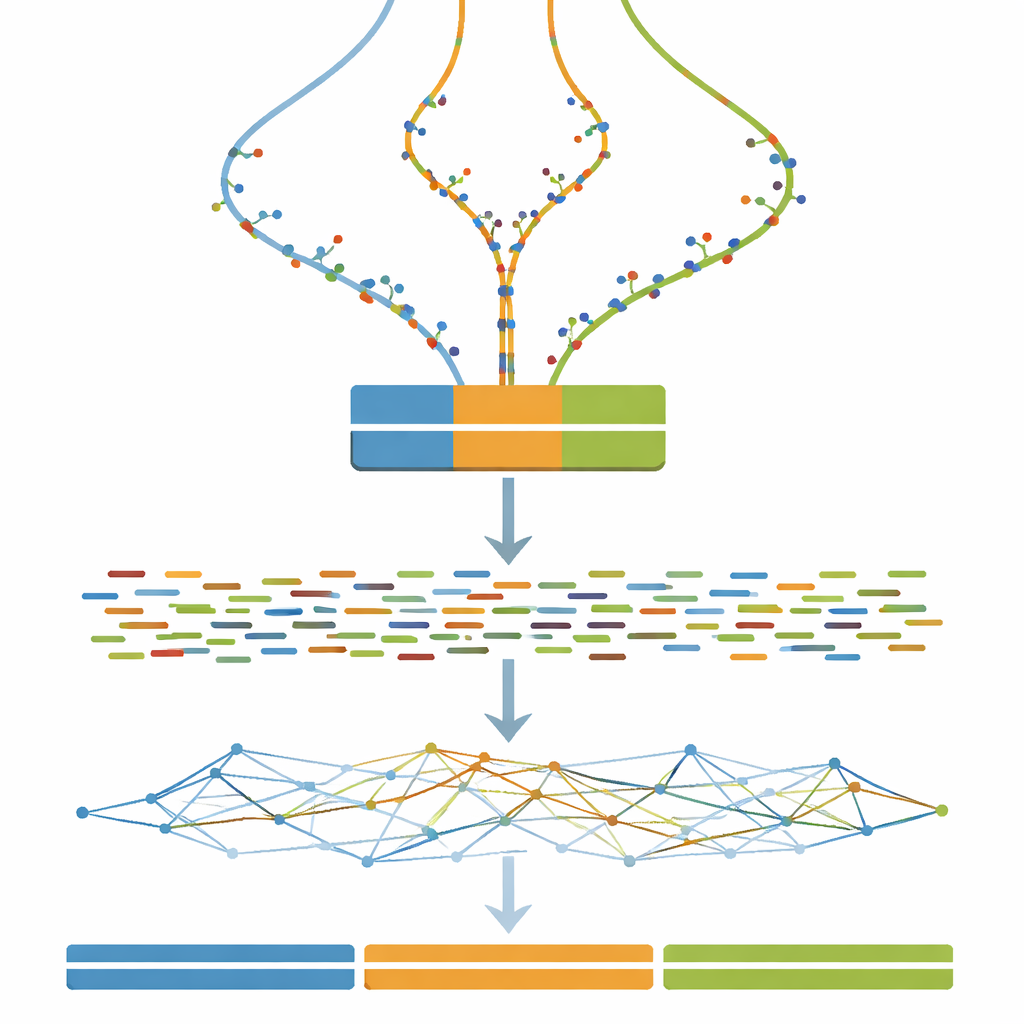
I stället för att utgå från en verklig växt eller ett djur skapar författaren först ett slumpmässigt DNA-avsnitt en miljon baspar långt som fungerar som en ren mall. Denna mall dupliceras sedan till tre separata versioner, som står för de tre kromosomuppsättningarna i en triploid organism. För att efterlikna hur verkliga genom förändras långsamt över tid inför studien ett fast antal små förändringar—enkelbas-substitutioner—steg för steg i varje kopia. Genom att upprepa denna process över 100 steg produceras tripletter av genom som varierar från nästan identiska till tydligt men ändå måttligt olika. Denna kontrollerade ”divergensgradient” utgör ryggraden i benchmarken.
Att förvandla virtuella genom till virtuella experiment
När varje trekopiersgenom är definierat är nästa steg att imitera vad en DNA-sekvenseringsmaskin skulle se. Studien använder väl etablerad programvara för att simulera korta parade DNA-fragment, liknande dem som produceras av en Illumina-sekvenserare, vid en konstant och ganska hög täckningsdjup. Valfria städatrinn efterliknar vanliga praktiker i verkliga tillämpningar, som att korrigera slumpmässiga sekvenseringsfel och slå ihop överlappande läspar. Som ett resultat kan vem som helst som använder datasetet testa inte bara sina assemblyalgoritmer utan också hur typiska förbehandlingsval påverkar de slutliga sammanställda genomen.
Stresstesta assemblystrategier
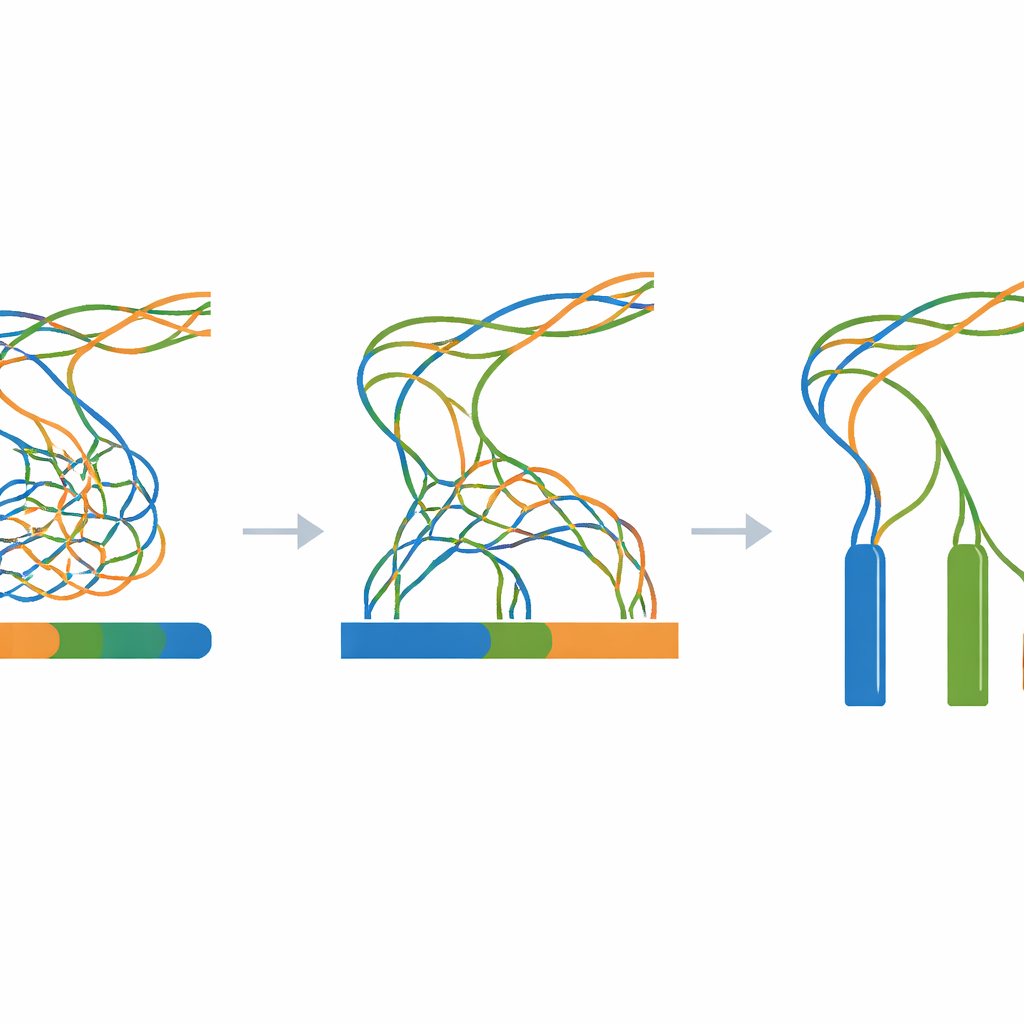
Kärnan i arbetet är ett omfattande experiment där alla simulerade läsningar matas in i ett enda genomsamlingsprogram medan man ändrar endast en viktig inställning: k-mer-storleken, en parameter som styr hur fint programvaran ”bitar upp” läsningarna när genomet rekonstrueras. För varje kombination av divergensnivå (från 0 till 100 steg) och k-mer-storlek (ett brett intervall av udda värden) byggs en ny assembly. Ett medföljande utvärderingsverktyg mäter sedan hur kontinuerliga de ihopbyggda fragmenten är, hur många fragment som finns och hur väl deras sammanlagda längd överensstämmer med den kända tre miljoner baspar långa sanningen. Dessa mätningar sammanfattas som värmekartor och avslöjar breda zoner där assemblyn kollapsar olika kopior till en, fragmenteras i många små delar eller närmar sig idealet med tre långa, korrekta contigs.
En transparent referens för svåra genom
Eftersom varje steg är syntetiskt och skriptat—from den initiala slumpmässiga mallen till de slutliga assemblyerna—kan forskare reproducera hela arbetsflödet på vilken standard-Linuxdator som helst med endast öppen källkodsprogramvara. Zenodo-arkivet som länkas i artikeln innehåller mallgenomet, alla mellanliggande muterade sekvenser, alla simulerade läsningar och varje assemblyresultat, tillsammans med loggar och enkla hjälpskript. Tekniska kontroller bekräftar att mutationsprocessen beter sig som förväntat, att de simulerade läsningarna matchar de begärda längderna och täckningen, och att assemblyerna visar det förväntade mönstret: kraftig överkollaps när de tre kopiorna är nästan identiska, och tydligare separation när de driver längre isär.
Vad detta betyder i klarspråk
I vardagliga termer erbjuder denna artikel en kontrollerad testbana för programvara som försöker återskapa tre liknande instruktionsböcker från högar av blandade fragment. Genom att gradvis öka hur olika de tre böckerna är, och genom att systematiskt ändra en nyckelinställning i återbyggnadsprocessen, gör datasetet det enkelt att se när och hur nuvarande metoder misslyckas eller lyckas. Utvecklare kan använda det för att finjustera nya algoritmer, medan användare kan bättre förstå vilka inställningar som fungerar bäst för triploida genom. Även om DNA:t självt är konstgjort är de insikter det möjliggör—om kollaps, separation och effekten av parameterval—direkt relevanta för verkliga ansträngningar att avkoda de komplexa genomerna hos många viktiga arter.
Citering: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Nyckelord: triploid genomassembly, polyploid benchmarking, syntetiskt DNA-dataset, de novo-assembly, k-mer-optimering