Clear Sky Science · sv
Föra proteomikdata bort från digitala gravgårdar genom gemensamt ansvar och samhällsengagemang
Varför dina medicinska data inte bör hamna i en digital gravplats
Modern medicin förlitar sig i allt högre grad på enorma datamängder som beskriver de tusentals proteiner som verkar i våra celler. Dessa filer delas ofta öppet online med löftet att andra forskare ska kunna dubbelkolla resultat eller ställa nya frågor utan att behöva göra nya experiment. Men om data publiceras i förvirrande format, saknar viktiga detaljer eller är bundna till proprietär programvara blir de till ”datagravgårdar”: synliga för alla men praktiskt taget oanvändbara. Den här artikeln visar hur en universitetskurs förvandlade studenter till datadektektiver för att avslöja detta dolda problem — och föreslår enkla åtgärder som kan göra delade data verkligen återanvändbara.
Lära vetenskap genom att upprepa riktiga studier
Vid Helsingin universitet (University of Helsinki) fick doktorandstudenter i en proteomikkurs med masspektrometri i uppdrag att göra något ambitiöst: välja verkliga, offentligt tillgängliga proteindataset från en stor arkiv och försöka reproducera de publicerade resultaten. I små team laddade de ned sex projekt från ProteomeXchange‑nätverket, som värd för masspektrometriska resultat från många laboratorier världen över. Med en gemensam analysrutin i programmeringsspråket R följde studenterna i stora drag samma steg som de ursprungliga forskarna: identifiera proteiner, mäta deras mängd, städa upp data och testa vilka proteiner som förändras mellan tillstånd, till exempel sjuk kontra frisk vävnad.
Stora löften, saknade instruktioner
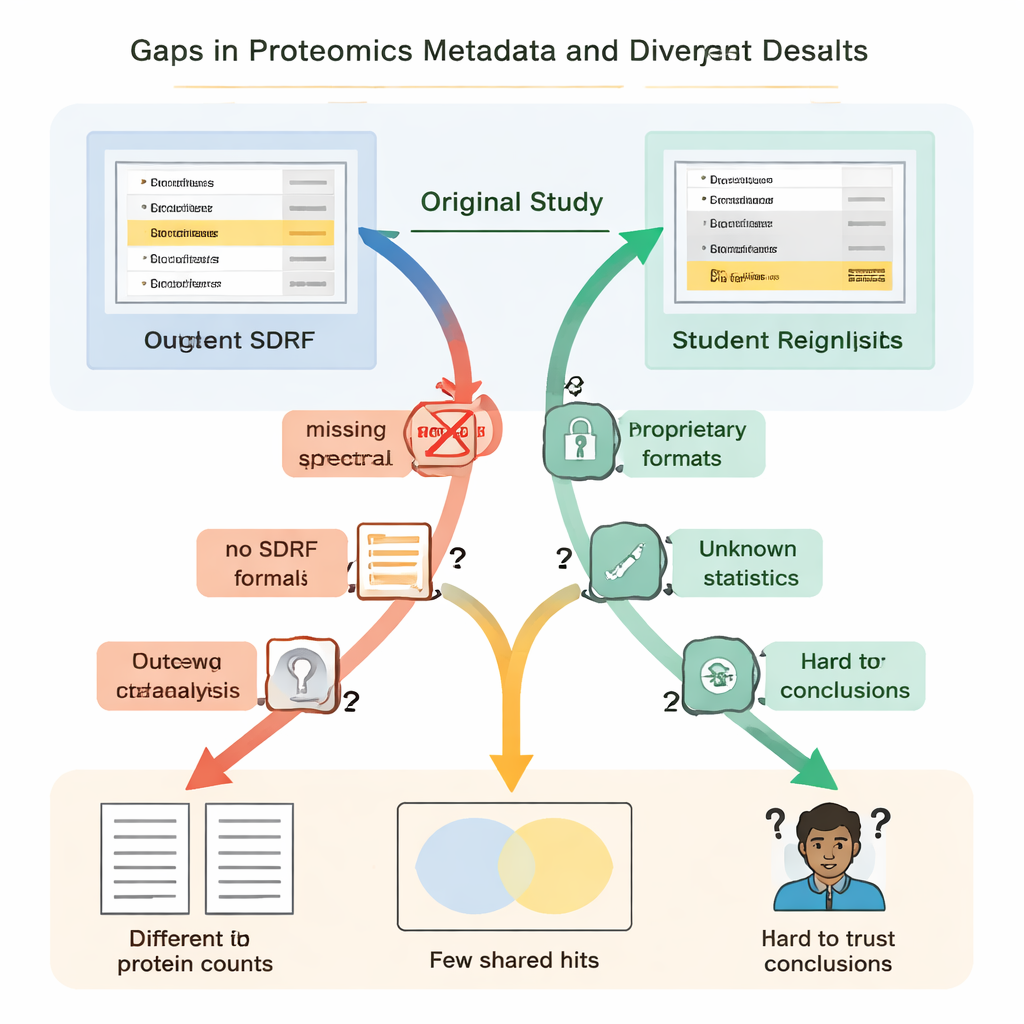
Studenterna upptäckte snabbt att ”öppet” inte alltid betydde ”återanvändbart”. I samtliga fall saknades väsentliga instruktioner eller var svåra att hitta. Viktiga länkar mellan prover och datafiler beskrevs inte i ett enkelt, maskinläsbart format, så teamen tvingades gissa vilka råfiler som hörde till vilka biologiska grupper genom att läsa artiklar och tyda filnamn. Uppgifter om hur falska positiva kontrollerades — till exempel användning av särskilda ”decoy” proteinsekvenser — saknades, vilket gjorde det omöjligt att noggrant bedöma hur trovärdiga de rapporterade proteinlistorna verkligen var. I flera projekt var huvudresultaten låsta i proprietära filformat eller beroende av kommersiell programvara som studenterna inte hade tillgång till, vilket tvingade dem att göra om stora delar av analysen från början.
När små luckor skapar stora skillnader
Dessa saknade detaljer var inte bara irriterande; de ledde till dramatiskt olika vetenskapliga resultat. I en njursjukdomsstudie rapporterade de ursprungliga författarna strax under femtusen proteiner, medan studenternas reanalys — med ett öppet verktyg och ett hembyggt spektralbibliotek — fann över tretton tusen. Ett protein som lyftes fram i originalartikeln som särskilt viktigt framträdde inte övertygande i den underliggande identifieringsfilen och upptäcktes inte alls i studenternas arbetsflöde. I ett annat fall listade originalstudien 108 proteiner som skiljde sig mellan tillstånden, men studenterna, som arbetade från samma rådata men med ofullständig information om hur originalstatistiken gjordes, kunde med tillförsikt flagga endast 11. Annorstädes gjorde avsaknaden av biologiska replikat i de uppladdade filerna att korrekt statistisk testning helt enkelt var omöjlig.
Vad ett ”återanvändbart” dataset egentligen bör innehålla
Från dessa sex fallstudier framträdde ett tydligt mönster: de största hindren för reproducerbarhet var inte masspektrometern i sig utan hur resultaten paketifierades och delades. Författarna argumenterar för att varje proteomikdataset bör levereras med ett minimalt re‑analys‑paket. Detta inkluderar rådata plus öppna, samhällsstandardiserade resultatformat; en standardiserad tabell som länkar varje prov till dess experimentella villkor; grundläggande kvalitetskontrollsammanfattningar; eventuella spektralbibliotek eller proteinsekvensfiler som behövs för att upprepa sökningen; samt fullständiga analysparametrar och kod, helst sparade med versionshanterade programvarukontainrar. Arkiv, tidskrifter och granskare kan hjälpa genom att uppmuntra eller kräva att inlämnare tillhandahåller detta paket från början, så att andra inte behöver rekonstruera arbetsflödet utifrån spridda ledtrådar.
Utbilda forskare samtidigt som systemet förbättras
Själva kursen fyllde ett dubbelt syfte. För studenterna erbjöd den ett praktiskt sätt att behärska komplexa proteomikmetoder, statistik och kodning, samtidigt som den visade hur sköra publicerade slutsatser kan vara när dokumentationen är ofullständig. För det bredare samhället gav studenternas kamp ett stresstest av rådande praxis för datadelning, och belyste exakt var metadata och analysregister brister. Författarna föreslår att liknande kurser kan köras på andra håll, och förvandla klassrum till kvalitetskontrollsmotorer som ständigt driver på för tydligare och mer transparenta data.
Från datagravgårdar till levande resurser
Enkelt uttryckt drar artikeln slutsatsen att många proteindataset som nu ligger i offentliga arkiv riskerar att bli digitala gravgårdar — kostsamma experiment vars resultat inte kan kontrolleras eller byggas vidare på på ett tillförlitligt sätt. Men lösningen är relativt enkel: behandla metadata, öppna format och delbar kod som integrerade delar av experimentet, inte som eftertankar. Om forskare, granskare och arkiv gemensamt insisterar på ett enkelt, väl dokumenterat paket när proteomikdata delas, kan dessa dataset förbli ”levande”: redo att reanalyseras, kombineras med nya studier och användas för att stärka bevisen bakom biomedicinska upptäckter.
Citering: Vadadokhau, U., Soliman, M., Castillon, L. et al. Preventing Proteomics Data Tombs Through Collective Responsibility and Community Engagement. Sci Data 13, 287 (2026). https://doi.org/10.1038/s41597-026-06614-8
Nyckelord: proteomik, datapålitlighet, öppen vetenskap, masspektrometri, delning av forskningsdata