Clear Sky Science · sv
AF2BIND: förutspår bindningsställen för småmolekyler med hjälp av AlphaFold2:s pair-representation
Att hitta läkemedelsmål i ett hav av proteiner
Moderna läkemedel verkar ofta genom att fästa vid små nischer på ytorna av proteiner inuti våra celler. Trots dagens enorma kataloger av proteinstrukturer är det ändå förvånansvärt svårt att i förväg avgöra var en småmolekyl – en potentiell läkemedelskandidat – faktiskt kan binda. Denna studie introducerar AF2BIND, ett enkelt men kraftfullt beräkningsverktyg som utnyttjar AlphaFold2:s inre representationer, den banbrytande proteinstrukturprediktorn, för att belysa sannolika läkemedelsbindningsställen över tusentals mänskliga proteiner. Målet är att begränsa sökfältet för nya läkemedel och avslöja dolda funktionella hot spots som traditionella metoder förbiser.
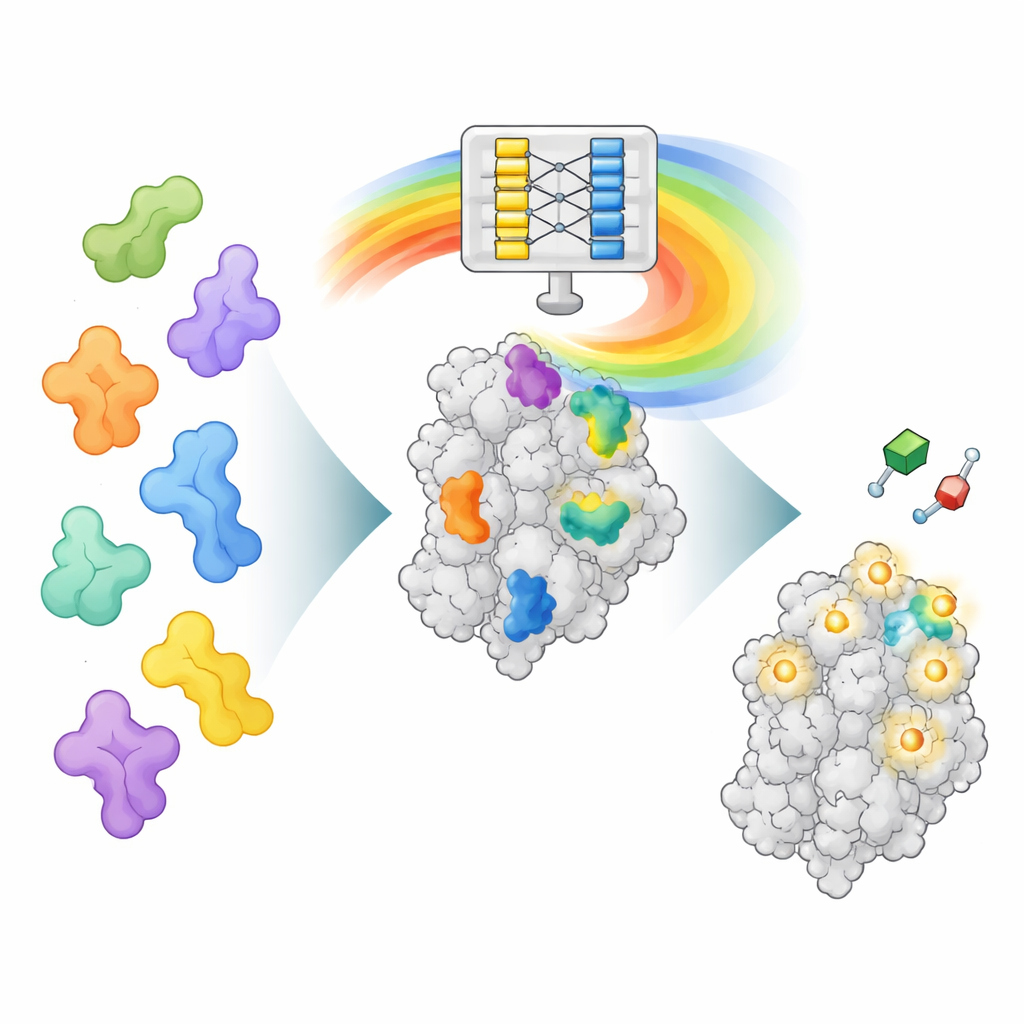
Ett nytt sätt att läsa AlphaFolds ”sinnelag”
AlphaFold2 tränades för att förutsäga hur en aminosyrakedja veckar sig till en tredimensionell proteinstruktur, inte för att hitta var läkemedel binder. Men i processen att lära sig vikningen fångade modellen också rika mönster om hur olika delar av proteiner interagerar. AF2BIND utnyttjar ett av dessa interna datalager, kallat pair-representation, som kodar hur varje par av aminosyrapositioner förhåller sig i rummet. Författarna matar AlphaFold2 med en proteinsekvens tillsammans med dess ryggradstruktur och lägger dessutom till 20 extra aminosyror, en av varje sort, som separata ”bete”-kedjor. AlphaFold2 beräknar sedan hur proteinet interagerar med varje beteresid. Dessa interaktionsmönster blir indata till en mycket enkel logistisk regressionsmodell som uppskattar, för varje position i proteinet, sannolikheten att den tillhör ett bindningsställe för småmolekyler.
Att omvandla dolda signaler till praktiska förutsägelser
Träningen av AF2BIND krävde ett noggrant kurerat set om cirka 1 900 protein–ligandstrukturer där småmolekyler var bundna med högkvalitativ experimentell evidens. Forskarna gick långt för att undvika ”fusk” via likhet: de delade upp sina data så att testproteiner inte delade övergripande fold, sekvens eller ens form av bindningsficka med de som användes för träning. På denna rigorösa referensprestanda överträffade AF2:s pair-representation flera alternativa neurala nätverksinbäddningar, inklusive sådana som enbart bygger på sekvens eller på strukturvillkorad sekvensdesign. Med enbart pair-funktionerna återfann AF2BIND ungefär två tredjedelar av kända bindande rester bland topp-rankade förutsägelser och visade stark prestanda över standardmått för klassificering, samtidigt som metoden var robust mot måttliga förändringar i proteinform och sidokedjeorienteringar.
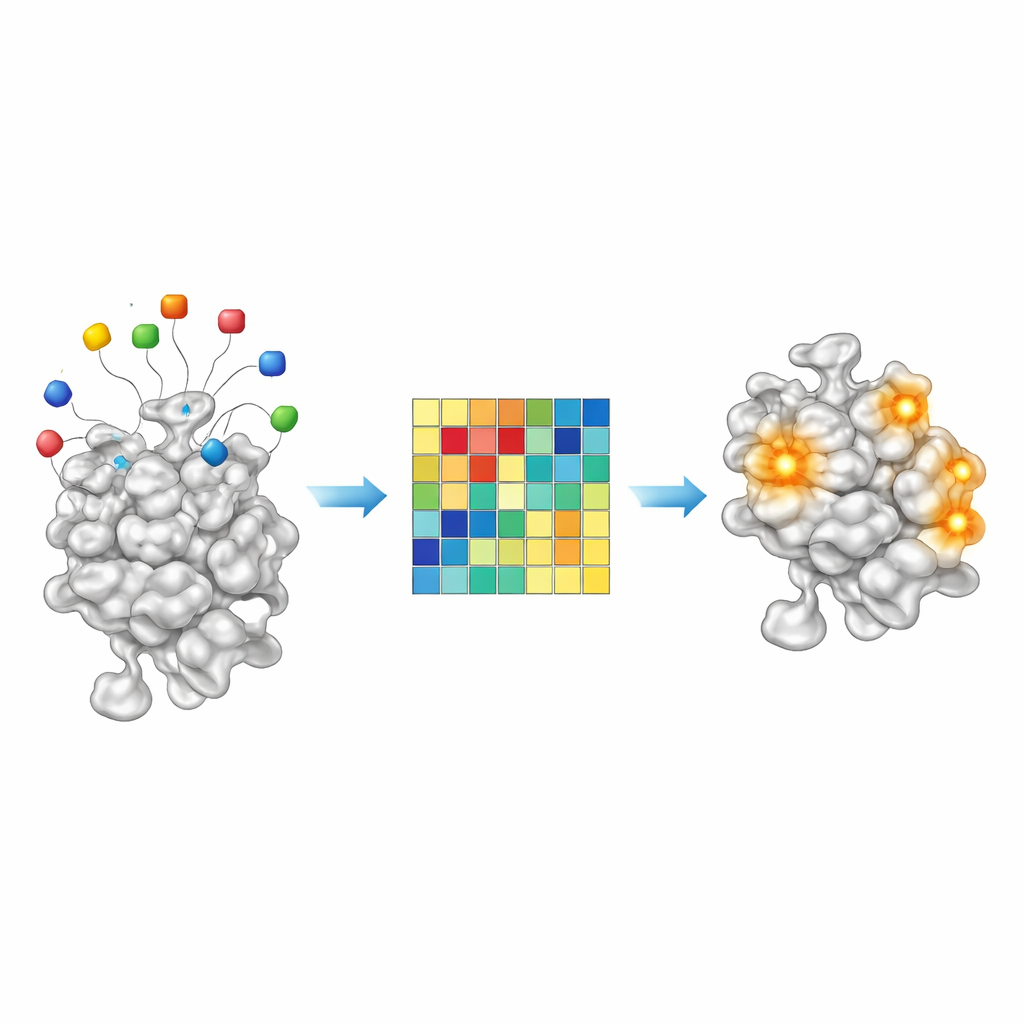
Läsa kemiska ledtrådar från bete-resid
Eftersom AF2BIND är en enkel linjär modell är dess beslut ovanligt transparenta för ett modernt AI-system. Var och en av de 20 bete-aminosyrorna bidrar med en mätbar mängd till det slutliga bindningsscoret vid en given proteinposition. Genom att undersöka dessa bidrag över cirka 2 000 protein–ligandkomplex fann författarna att vissa bete-kombinationer aktiverar starkare för oljiga, kolrika ligander, medan andra tänds för mer polära, vattenälskande molekyler. Med andra ord fungerar mönstret av bete-aktivering som ett grovt kemiskt fingeravtryck för vilka typer av småmolekyler en viss ficka föredrar. Detta antyder att AF2BIND-liknande angreppssätt i framtiden inte bara kan flagga var ett läkemedel kan binda, utan också ge ledtrådar om vilken sorts kemi som skulle passa bäst.
Skanning av den humana proteomen efter nya fickor
Utrustade med sin tränade modell släppte teamet sedan AF2BIND lös på AlphaFold-prediktionerna av hela den humana proteomen. Efter att ha trimmat bort regioner med låg förtroendenivå och delat mycket stora proteiner i hanterbara strukturella bitar, klustrade de närliggande högt rankade rester till kandidat-bindningsställen. AF2BIND förutsade över 20 000 sådana ställen i mer än 13 000 proteiner. Slående nog överlappade majoriteten av dessa inte med fickor härledda av homologi-baserade metoder som AlphaFill, som kopierar ligander från närliggande kristallstrukturer, heller inte med en mycket använd fickletare kallad P2Rank. Många AF2BIND-exklusiva ställen är grundare eller mer diffusa än klassiska begravda fickor och sammanfaller ofta med regioner som binder peptider, RNA, DNA eller andra proteiner—gränsytor som ändå kan vara riktbara med småmolekyler.
Konsekvenser för läkemedelsupptäckt och sjukdom
För att bedöma hur lovande dessa nyföreslagna ställen kan vara för läkemedelsdesign använde författarna ett oberoende verktyg som poängsätter ”druggability” baserat på fickstorlek, inneslutning och kemisk miljö. I genomsnitt poängsatte AF2BIND:s ställen över en vanlig tröskel för attraktiva läkemedelsmål, inklusive sådana som finns i proteiner kopplade till ärftliga sjukdomar. När resultaten korsrefererades med kemoproteomiska experiment som märker reaktiva cysteiner i celler förklarade AF2BIND och P2Rank tillsammans nästan hälften av de observerade ligandbara regionerna, där varje metod fångade fall som den andra missade. Arbetet visar att de interna representationer som lärts upp av strukturprediktionsnätverk kan återanvändas för att kartlägga sannolika läkemedelsbindningsställen i massiv skala, utan förkunskap om någon specifik ligand. För icke-specialister är huvudbudskapet att samma AI-genombrott som förutser proteinformer börjar avslöja var och hur läkemedel bäst kan greppa dessa former, vilket potentiellt kan snabba upp sökandet efter nya behandlingar och belysa tidigare dolda kontrollpunkter i våra proteiner.
Citering: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Nyckelord: proteinbindningsställen, läkemedelsupptäckt, AlphaFold2, beräkningsbiologi, strukturell bioinformatik