Clear Sky Science · sv
DECODE: djupinlärningsbaserat generellt dekonvolutionsramverk för olika omikdata
Varför denna forskning är viktig
Modern biomedicin översvämmas av mätningar från våra vävnader: vilka gener som är aktiva, vilka proteiner som finns och vilka småmolekyler som driver våra celler. De flesta av dessa mätningar görs dock på sammansatta prover där många celltyper blandas. Studien bakom DECODE introducerar ett kraftfullt ramverk baserat på artificiell intelligens som kan avblanda dessa signaler och berätta vilka celler och celltillstånd som finns närvarande, även över mycket olika datatyper. Denna förmåga kan påskynda forskning om cancer, immunitet och metabola sjukdomar samtidigt som den gör bättre användning av befintliga biobanksprover.
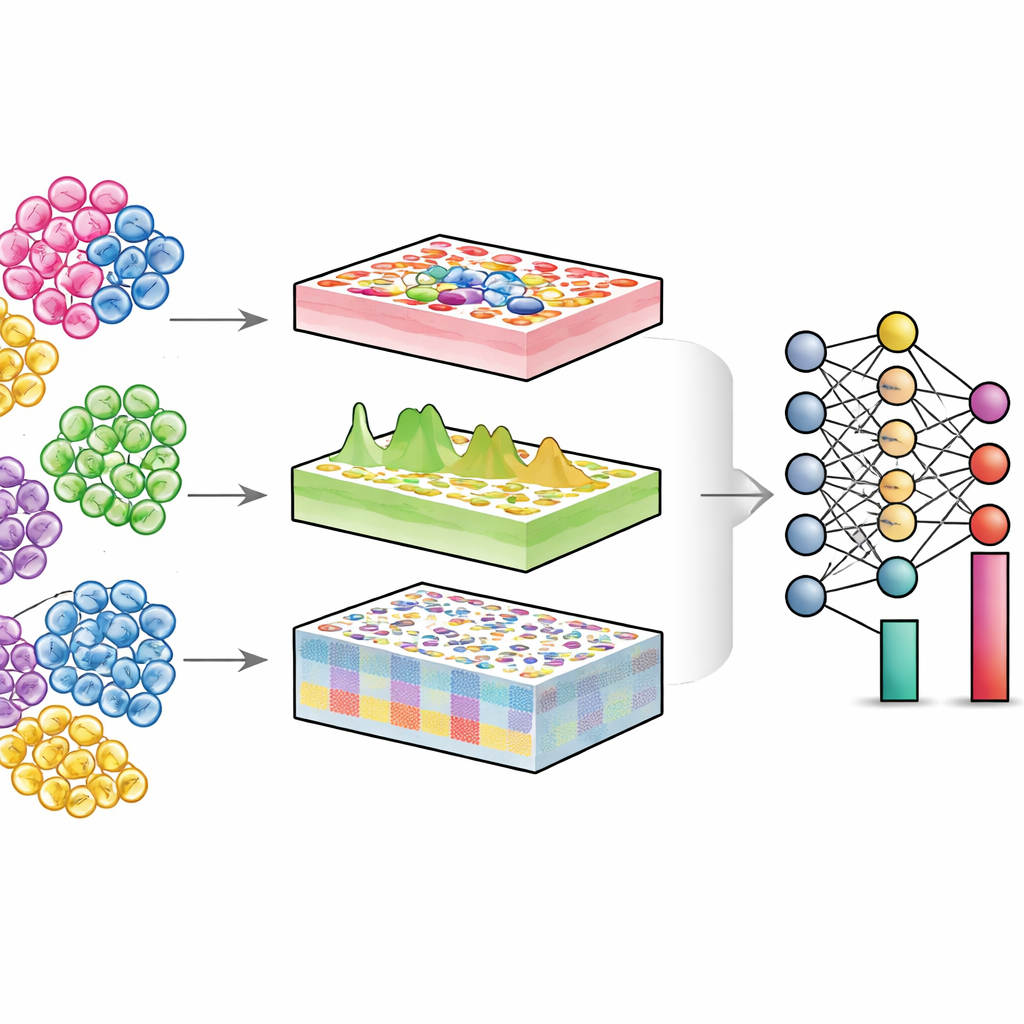
En titt in i blandade vävnader
Varje organ är ett samhälle av olika celltyper—immunceller, strukturella celler, stamceller och fler. Vid hälsa och sjukdom är det ofta inte bara vad varje cell gör som förändras, utan också hur många av varje sort som finns och i vilket tillstånd de befinner sig. Single-cell-teknologier kan mäta individuella celler direkt, men de är dyra och tekniskt krävande, särskilt för stora patientkohorter eller äldre lagrade prover. I kontrast blandar konventionella ”bulk”-experiment tusentals eller miljoner celler och läser av en genomsnittssignal. Dekonvolutionsalgoritmer försöker vända denna blandning: givet bulkdata och en referenskarta av single-cell-data estimerar de andelen av varje celltyp i vävnaden.
Begränsningar hos specialiserade verktyg
Befintliga dekonvolutionsverktyg är mestadels anpassade till en enda mätmetod, såsom genaktivitet (transkriptomik) eller proteiner (proteomik). De antar ofta specifika statistiska beteenden som inte gäller för andra datatyper, och de har svårt när den bulkvävnad som analyseras innehåller celltyper som saknas i referensen. Starka batcheffekter—skillnader i donatorer, instrument eller hälsotillstånd—kan dessutom sudda ut de biologiska signalerna. Noterbart är att det saknades en praktisk metod för metabolomik, studiet av små molekyler som ofta ligger närmast kliniska symptom. Som en följd tvingades forskare som analyserade multiomiska kohorter hantera flera specialverktyg, vardera med egna egenheter, vilket försvårade jämförelser mellan studier och datatyper.
En universell avblandningsmotor
DECODE tar itu med dessa utmaningar genom att betrakta dekonvolution som ett flexibelt djupinlärningsproblem som kan hantera gener, proteiner och metaboliter på ett enhetligt sätt. Först syntetiserar det ”pseudovävnader” genom att digitalt blanda single-cell-profiler i slumpmässiga proportioner och skapar därigenom en rik träningsuppsättning där den verkliga cellkompositionen är känd. Ett adversarialt inlärningssteg lär sedan en encoder att avbilda både riktiga vävnader och pseudovävnader till en gemensam representation där tekniska skillnader minimeras men biologiskt meningsfulla mönster bevaras. Därefter lär ett särskilt avbrusningsmodul, vägledd av kontrastiv inlärning, att separera verkliga vävnadssignaler från artificiellt brus. Detta steg gör DECODE robust mot saknade celltyper i referensdata och mot mätfel. Slutligen skickas de renade kännetecknen till en dekonvolutionsmodul som estimerar antingen absoluta eller relativa abundanser av celltyper och celltillstånd, beroende på hur komplett referensen är.
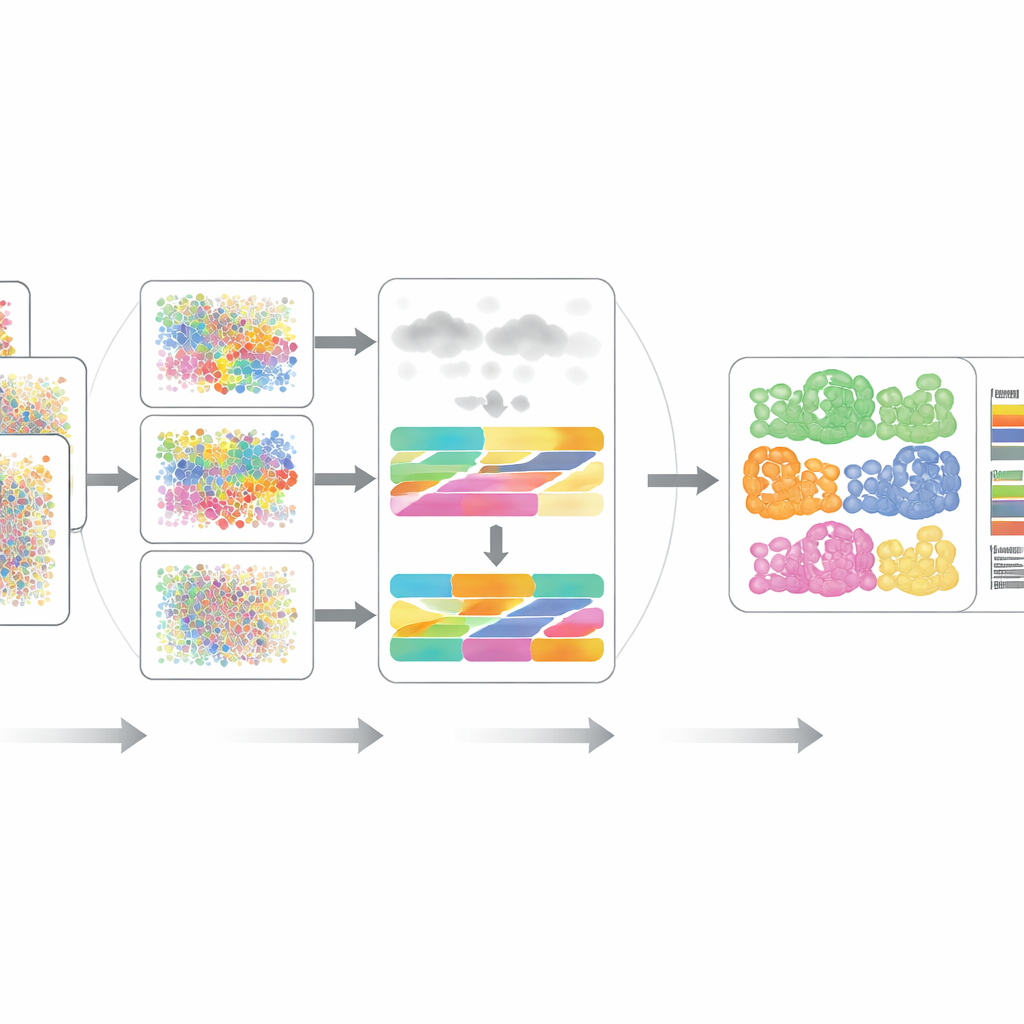
Att pröva DECODE
Författarna benchmarkade noggrant DECODE på 15 dataset som täcker sju realistiska scenarier, inklusive olika donatorer, sjukdomstillstånd, hälsotillstånd, experimentella plattformar och till och med rumsligt upplösta mätningar. Över transkriptomik och proteomik matchade eller överträffade DECODE generellt topptjänsterna vad gäller noggrannhet samtidigt som rimlig beräkningstid och minnesanvändning upprätthölls. Avgörande var att DECODE var den enda metoden som levererade tillförlitliga resultat på metabolomikdata, där det finns färre funktioner och olika celltyper kan se bedrägligt lika ut. Ramverket visade sig också skickligt på att följa celltillstånd—såsom progression längs en utvecklingstrajektoria, faser i cellcykeln eller svar på läkemedelsbehandling—snarare än bara statiska celltyper.
Robust i brusiga och ofullständiga verkliga data
Riktiga vävnader innehåller ofta celltyper som inte fångas i labb-baserade single-cell-referenser, och experimentellt brus kan förvränga många funktioner samtidigt. Forskarna simulerade dessa problem genom att lägga till okända celltyper och introducera flera varianter av brus och saknade data över transkriptomik, proteomik och metabolomik. I de flesta inställningar förblev DECODE den mest precisa metoden och, inom metabolomik, den enda som inte kollapsade. De visade också att DECODE ger mycket konsekventa svar när det tillämpas på matchade gen- och proteinmätningar från samma blodcellprover—ett nyckelkrav för att jämföra celltypförändringar över omiklager i stora kohorter.
Nya biologiska insikter från multiomiska kohorter
Med detta enhetliga verktyg återbesökte teamet komplexa sjukdomsdatamängder. Vid bröstcancer kombinerade de transcriptomiska och proteomiska kohorter för att visa hur immunceller och stödjande stromaceller skiftar mellan icke‑metastatiska tumörer, metastaserande primära tumörer och hjärnmetastaser. Mönster såsom högre förekomst av T‑celler och perivaskulära liknande celler i icke‑metastatiska lesioner, och ökade B‑celler i avancerad sjukdom, stämmer överens med och utvidgar tidigare biologiska studier. I muslever integrerade DECODE transkriptomiska, proteomiska och metabolomiska kohorter för att följa hur hepatocyter, endotelceller och residenta immunceller förändras vid olika dieter och levermodeller, och återgav kända trender som ökande andel Kupffer-celler vid inflammatoriska tillstånd.
Vad detta betyder framöver
För en lekmannaläsare är huvudbudskapet att DECODE fungerar som ett smart prisma för biomedicinska data: givet blandade mätningar från vävnader kan det separera bidragen från många olika celltyper och tillstånd, och det gör det på ett tillförlitligt sätt över flera typer av molekylära avläsningar. Detta gör det möjligt för forskare att pressa mycket mer information ur befintliga multiomiska kohorter och biobanker utan att samla in nya single-cell-data för varje projekt. Även om metoden fortfarande är beroende av kvaliteten och bredden hos tillgängliga single-cell-referenser, och resurser inom metabolomik förblir begränsade, markerar DECODE ett betydande steg mot rutinmässig tolkning på cellnivå av storskaliga humanstudier, med potentiella fördelar för att förstå sjukdomsmekanismer och vägleda precisionsmedicin.
Citering: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Nyckelord: multiomisk dekonvolution, single-cell-referens, djupinlärning inom biologi, metabolomikanalys, celltypkomposition