Clear Sky Science · sv
Hastighetsvariation och återkommande sekvensfel i pandemiskala fylogenetiska studier
Varför detta spelar roll vid framtida utbrott
När ett nytt virus sprider sig över världen tävlar forskare om att avläsa dess genetiska kod och återskapa dess släktträd. Dessa träd hjälper till att spåra hur varianter uppstår, hur snabbt de sprids och om kontrollåtgärder fungerar. Men under COVID-19 sekvenserade laboratorier miljoner SARS‑CoV‑2-genom så snabbt att dolda fel och egenheter i data började förvränga bilden. Denna artikel presenterar nya metoder för att rensa och tolka sådana omfattande genetiska datamängder och erbjuder klarare insikter i hur ett pandemivirus verkligen utvecklas och rör sig i populationer.
Utmaningen i att tolka miljontals genom
Genomisk epidemiologi omvandlar virusgenom till praktisk information för folkhälsobeslut. För SARS‑CoV‑2 har mer än 20 miljoner genom delats globalt. Traditionella evolutionära verktyg byggdes för mer blygsamma problem, som att jämföra gener mellan arter, inte för att hantera miljontals nästan identiska virussekvenser som anländer i realtid. I denna skala blir två problem särskilt besvärliga. För det första muterar vissa positioner i virusgenomet mycket oftare än andra, vilket kan få icke‑besläktade virus att verka onormalt lika. För det andra kan återkommande tekniska fel i sekvensering och databehandling efterlikna verkliga mutationer. Båda effekterna skapar ”falska ekon” i det evolutionära trädet och ökar osäkerheten kring vilka grenar och grupperingarna man kan lita på.
Att upptäcka snabbföränderliga positioner och dolda misstag
Författarna utökar sitt fylogenetiska program, MAPLE, med modeller som behandlar varje position i virusgenomet som om den hade sitt eget beteende. Istället för att anta ett fåtal genomsnittliga mutationshastigheter uppskattar metoden en separat hastighet för varje site och utnyttjar det stora antalet tillgängliga genom. Samtidigt tillåter den att varje site har en egen sannolikhet för att bära ett återkommande sekvenserings‑ eller konsensus‑fel. Huvudtricket är att jämföra hur ofta en förändring dyker upp på djupa interna grenar i trädet, som speglar äldre, delade händelser, jämfört med de yttersta spetsarna, som motsvarar individuella genom. Sanna biologiska mutationer tenderar att vara fördelade mellan interna och terminala grenar, medan tekniska fel visar sig främst vid spetsarna. Genom att utnyttja detta mönster kan metoden skilja verkligt snabb evolution från återkommande fel.
Snabbare algoritmer för ett trångt trä av livet
Att hantera miljontals genom skulle normalt kräva enorma beräkningsresurser. För att hålla analysen praktisk redesignade teamet hur MAPLE lagrar och uppdaterar sekvensinformation i trädet. Istället för att jämföra varje genom med en enda fast referens väljer programvaran ”lokala referenspunkter” inuti trädet och registrerar närliggande genom som skillnader relativt dessa ankare. Denna kompakta representation snabbar upp jämförelser mellan avlägsna delar av trädet. Ytterligare förbättringar förfinar hur nya prover läggs till i ett befintligt träd, hur grenlängder justeras och hur sannolika alternativa trädformer utforskas, med möjligheter att köra de mest krävande stegen parallellt över flera processorkärnor.
Testning av metoden och rensning av verkliga data
För att kontrollera att deras modeller fungerar skapade författarna först realistiska simulerade SARS‑CoV‑2-datasets med kända mutationsmönster och inbäddade sekvensfel. I dessa tester återfann den nya metoden sannare evolutionära träd och lokaliserade enskilda fel med hög precision, särskilt när tiotusentals genom eller fler ingick. De vände sig sedan till verkliga data och analyserade miljontals SARS‑CoV‑2-sekvenser för vilka råa läsningar fanns tillgängliga. Genom att jämföra två olika konsensus‑byggande pipelines identifierade de specifika genompositioner som upprepade gånger påverkades av artefakter, såsom primerbindningsproblem eller referens‑bias i kallning. Dessa misstänkta positioner maskades från vidare analys, och genom som visade tecken på kontaminering eller blandinfektion filtrerades bort, vilket resulterade i en kurerad allians med över två miljoner högkvalitativa sekvenser.
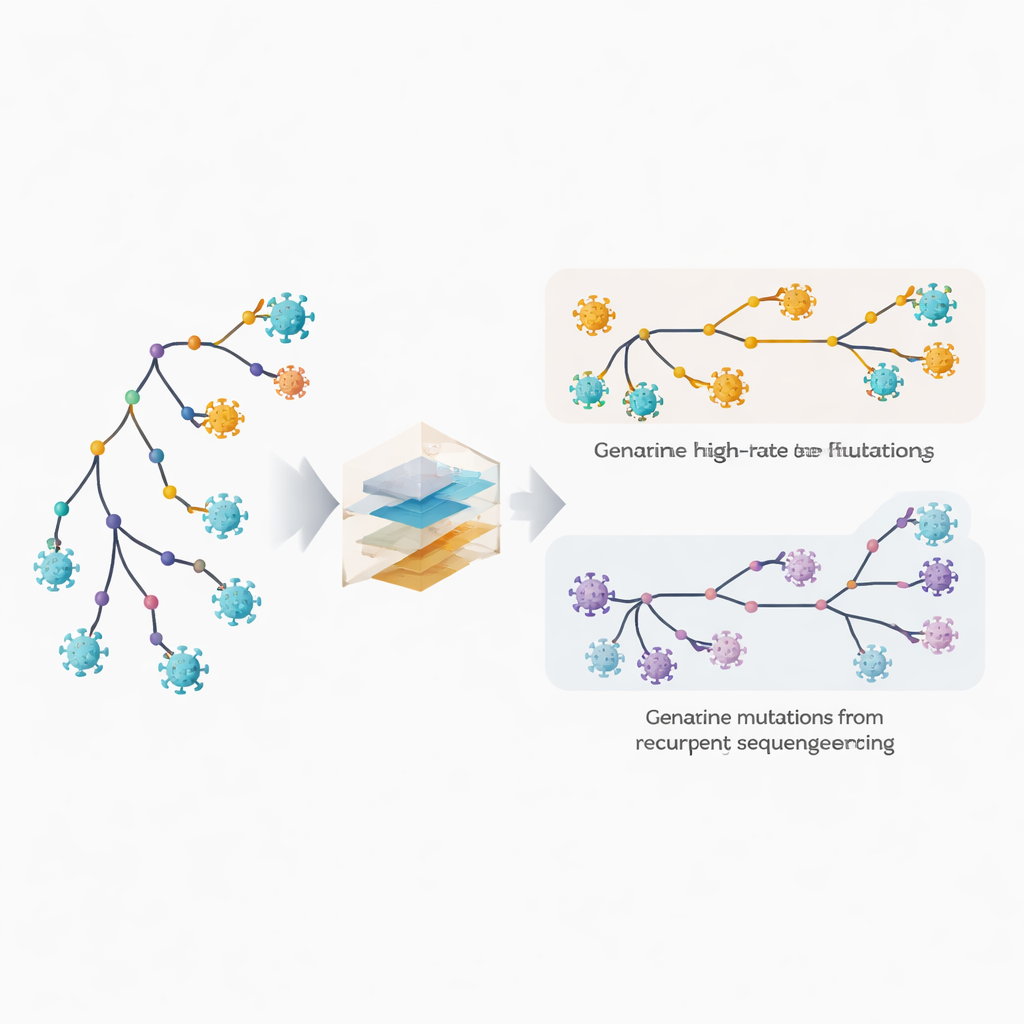
En klarare global bild av virusets släktträd
Med den rensade datamängden återskapade författarna ett globalt fylogenetiskt träd för SARS‑CoV‑2 och kartlade hur stora varianter förhåller sig till varandra. Deras träd föreslår ibland subtilt annorlunda relationer än tidigare publika träd, ofta på ett sätt som kräver färre mutationer och bättre överensstämmer med den statistiska modellen. Ramverket lyfter också fram var linjenamn kan vara inkonsekventa med den underliggande genetiska historien och flaggar möjliga rekombinanter eller problematiska genom för närmare granskning. Även om vissa utmaningar återstår — som överanpassning när data är knappa eller påverkan från starkt kontaminerade prover — visar arbetet att det nu är genomförbart att bygga mer tillförlitliga evolutionära träd i pandemiskala. För en allmän läsare är slutsatsen att bättre hantering av fel och mutationshotspots ger skarpare insikt i hur patogener sprids och förändras, vilket hjälper forskare och hälsomyndigheter att reagera snabbare och mer säkert vid framtida utbrott.
Citering: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Nyckelord: SARS-CoV-2-genomik, fylogenetiska metoder, sekvenseringsfel, variation i mutationshastighet, genomisk epidemiologi