Clear Sky Science · sv
Överförbara enantioselektivitetsmodeller från glesa data
En smartare väg för att hitta rätt katalysator
Kemister söker ofta efter bättre läkemedel och material genom att försöka föra samman kolatomer i mycket specifika tredimensionella arrangemang. Att uppnå det subtila «högerhänta» kontra «vänsterhänta» utfallet—kallat enantioselektivitet—innebär vanligtvis att man provar många metallkatalysatorer och reaktionsbetingelser genom försök och fel. Denna artikel presenterar ett sätt att använda relativt små mängder experimentella data, kombinerat med snabba datorberäkningar, för att förutsäga vilka nickelbaserade katalysatorer som ger önskad handningspreferens i ett brett spektrum av reaktioner och därigenom potentiellt spara kemister veckor eller månader av laboratoriearbete.
Varför handade molekyler är så svåra att kontrollera
Många läkemedel och naturliga produkter förekommer i spegelbildsformer som kan bete sig mycket olika i kroppen. Katalysatorer som gynnar den ena spegelbilden framför den andra är därför mycket värdefulla. Men att designa sådana katalysatorer är komplicerat. Traditionell kvantkemi kan i princip beräkna vilken reaktionsväg som föredras, men små energifel översätts till stora misstag i förutsagd selektivitet, och beräkningarna är långsamma. Enklare statistiska modeller är å andra sidan snabba men bortser ofta från den detaljerade interaktionen mellan metallen och reagerande molekyler, särskilt när reaktionsmekanismen kan förändras subtilt beroende på vilka partners som används.
Fånga de viktiga ögonblicken i en reaktion
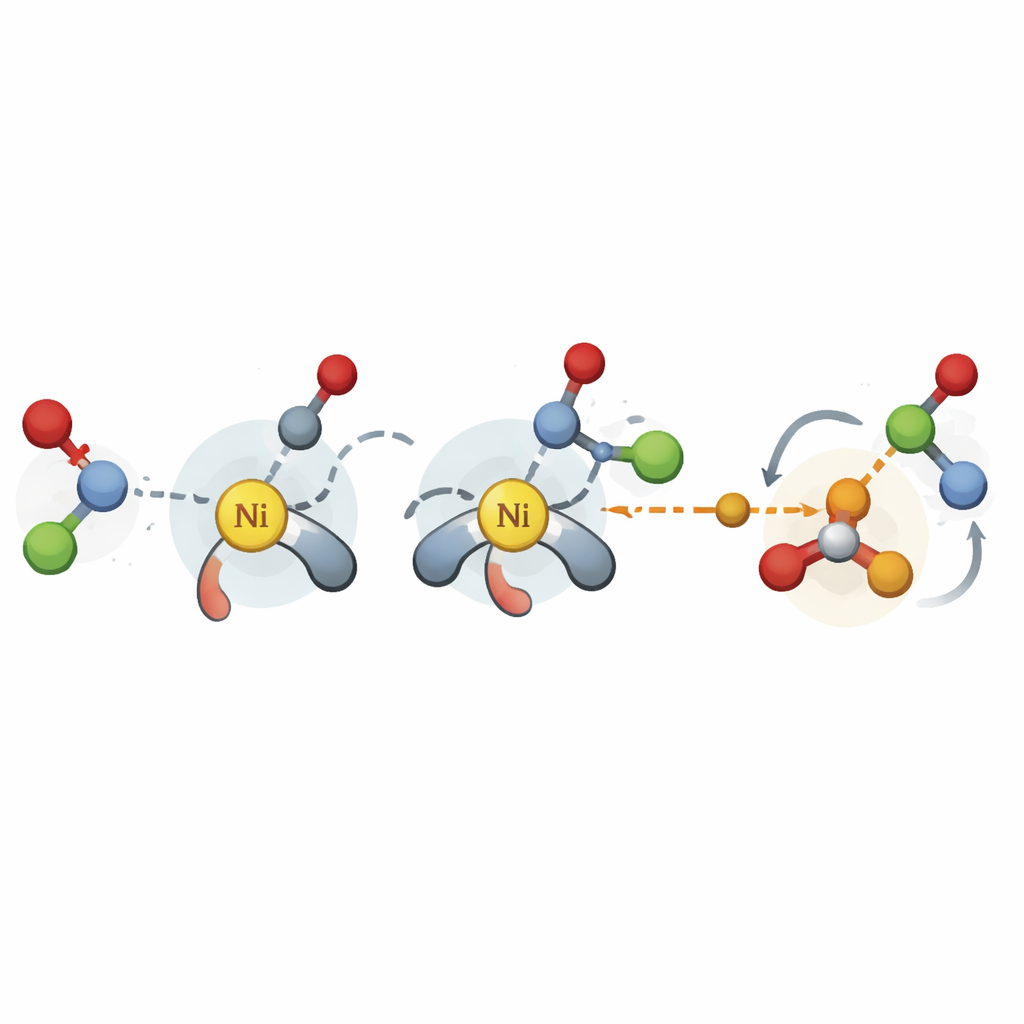
Författarna överbryggar denna klyfta genom att fokusera på de mest kritiska stegen i en nickelkatalyserad korskopplingsreaktion: stegen där nya kol–kol-bindningar bildas och produktens frigörande. Istället för att köra dyra högkvalitativa simuleringar använder de en effektiviserad kvantmetod för att generera tredimensionella strukturer för nyckelövergångstillstånd och intermediärer över många möjliga katalysator- och substratkombinationer. Från dessa strukturer extraherar de hundratals fysikaliskt meningsfulla deskriptorer, såsom hur trångt det är i katalysatormiljön nära vissa atomer eller hur lätt elektroner kan förflytta sig. Dessa tal matas sedan in i enkla linjära regressionsmodeller som kopplar strukturella egenskaper till uppmätt selektivitet.
Lära från glesa data för att styra nya experiment
Ett centralt resultat i arbetet är att det utnyttjar glesa data—de begränsade kombinationer av katalysatorer och substrat som typiskt rapporteras i en forskningsartikel—på bästa sätt. I en fallstudie återbesöker teamet en nickelreaktion som kopplar styrenoxider med aryljodider. De visar att deskriptorer tagna från det mest relevanta övergångstillståndet överträffar dem från förenklade katalysatorfragment, även om de bakomliggande beräkningarna är billigare. Med dessa modeller i hand testar de virtuellt många fler ligander på befintliga substratpar och identifierar nya katalysatorval som höjer den enantiomeriska överskottet för särskilt svårlösta exempel, samtidigt som de undviker dussintals onödiga experiment.
Överföra kunskap mellan olika reaktioner
Metoden är kraftfull eftersom den kan överföras mellan olika, men besläktade, nickelkatalyserade reaktioner. I en andra serie studier kombinerar författarna data från flera typer av nickelreaktioner som alla bildar bindningar mellan sp3-hybridiserade kolatomer och partners som aryl- eller alkenylgrupper, även när exakta betingelser eller kopplingspartners skiljer sig åt. Genom att bygga modeller från samma mekanistiskt meningsfulla deskriptorer förutsäger de framgångsrikt enantioselektivitet för nya ligander, nya substratkombinationer och till och med en helt ny klass av kol–kol-bindningsbildande reaktion som inte ingick i träningsdata. Analys av vilka deskriptorer som är mest betydelsefulla ger också ledtrådar om vilket steg i katalyscykeln som faktiskt bestämmer handningspreferensen för varje reaktionsfamilj.
Hjälpa kemister att starta nya reaktioner snabbare
I en avslutande demonstration använder författarna sitt deskriptorschema tillsammans med en bayesiansk optimeringsplattform för att designa en nickelkatalyserad koppling mellan benzylic acetaler och aryljodider som tidigare inte utvecklats asymmetriskt. Genom att utgå från litteraturdata om andra reaktioner rekommenderar modellen små satser lovande ligander att testa och hittar snabbt den bäst presterande klassen på bara några dussin experiment. För en kemist innebär detta ett praktiskt verktyg för att »kallstarta» ett nytt katalytiskt projekt: genom att mata in ett fåtal tidiga resultat kan modellen föreslå vilka chirala ligander som sannolikt ger hög enantioselektivitet. Sammanfattningsvis visar studien att väl utvalda, lågkostnadsberäknade funktioner kan omvandla begränsade tidigare data till allmänt användbara riktlinjer för att bygga nästa generation av selektiva reaktioner.
Citering: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Nyckelord: asymmetrisk katalys, nickel-kopplingsreaktion, maskininlärning i kemi, reaktionsoptimering, prediktion av enantioselektivitet