Clear Sky Science · sv
Bidirektionella CRISPR-skärmar avkodar en GLIS3-beroende fibrotisk cellkrets
När läkning blir skadlig ärrbildning
Våra tarmar är avsedda att reparera sig efter varje skada och irritation. Men vid kroniska sjukdomar som Crohns sjukdom och ulcerös kolit kan denna läkningsprocess spåra ur och leda till tjock, styv ärrvävnad som förtränger tarmen och kan kräva kirurgi. Denna studie avslöjar en dold konversation mellan immunceller och strukturella celler i tarmen som driver denna ärrbildning, och pekar ut en huvudbrytare, genen GLIS3, som kan erbjuda ett nytt sätt att bryta cykeln.
Ett dolt nätverk i inflammerade tarmar
För att förstå varför vissa patienter utvecklar envis inflammation och fibros skapade forskarna en cellulär ”atlas” över människotarmen. De kombinerade enkelcells-RNA-sekvensering, som läser av vilka gener som är aktiva i enskilda celler, med rumslig profilering som kartlägger var dessa celler sitter i riktiga vävnadssnitt. Med prover från personer med Crohns sjukdom, ulcerös kolit och kontroller kartlade de mer än fyra miljoner celler genom tarmväggen. Bland denna folkmassa framträdde en fibroblastundergrupp: inflammationsassocierade fibroblaster, eller IAFs. Dessa celler samlades i områden med aktiv och kronisk kolit och bar en gendetalj kopplad till resistens mot vanliga anti‑TNF‑behandlingar, vilket antyder att de spelar en central roll i svårbehandlade former av sjukdomen. 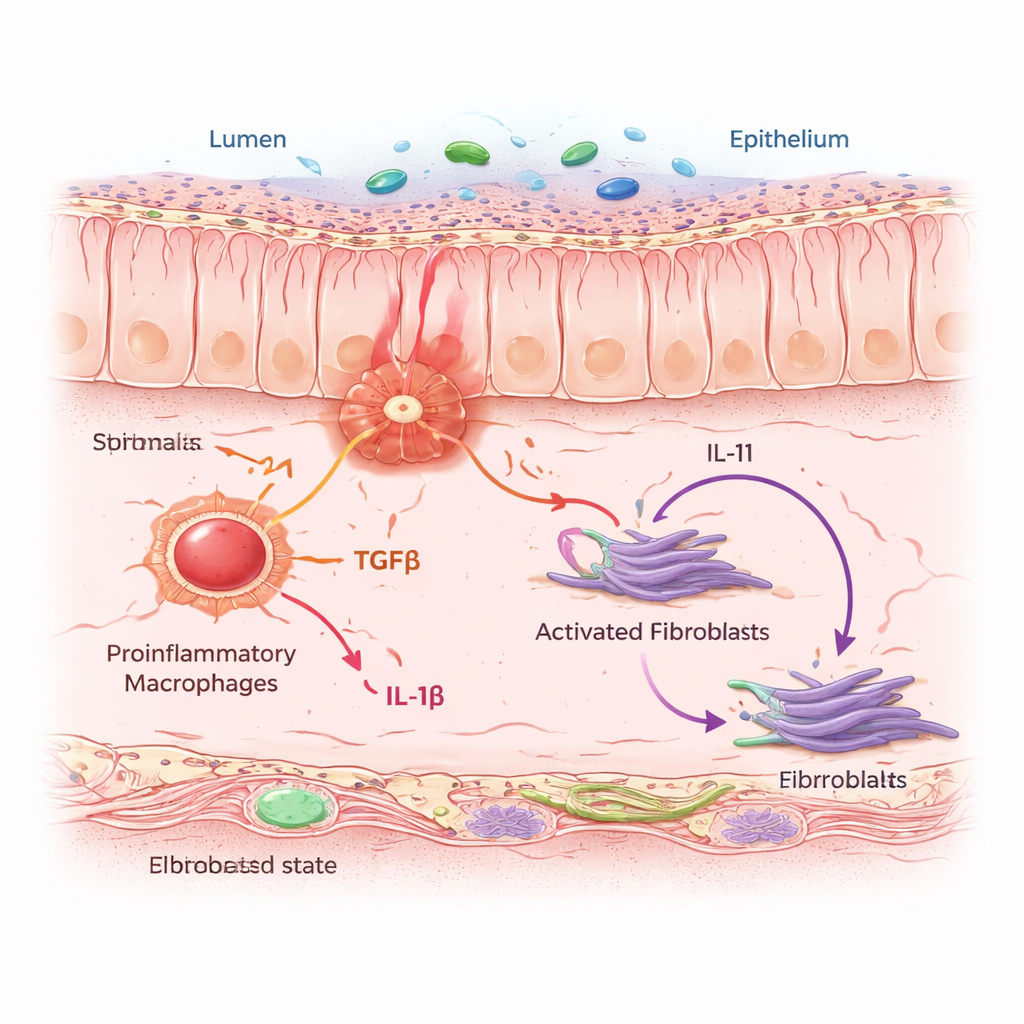
Makrofager viskar, fibroblaster skapar ärr
IAFs agerade inte ensamma. De klustrade i ”grannskap” täta med proinflammatoriska makrofager—immunceller som känner av fara och skickar ut larm. Genom beräkningsmodeller och cellko-kulturexperiment visade teamet att när makrofager drivs in i ett inflammatoriskt tillstånd utsöndrar de två viktiga budbärarproteiner: TGFβ och IL‑1β. Närliggande fibroblaster ”lyssnar” efter dessa signaler via specifika receptorer. När båda signalerna anländer samtidigt omkopplar fibroblasterna till IAF‑tillståndet och börjar producera IL‑11, en cytokin som redan misstänks främja fibros, tillsammans med kollagen och andra matrixproteiner som förtjockar och stelnar tarmväggen. Hos möss utsatta för ett kroniskt kolit‑regimen minskade blockerande av IL‑11 eller selektiv borttagning i fibroblaster kollagenansamling utan att hindra den initiala inflammationen, vilket visar att IL‑11 är en avgörande drivkraft i ärrbildningsfasen.
GLIS3: huvudbrytaren i fibrotiska fibroblaster
För att gå från korrelationer till mekanismer använde författarna kraftfulla genomsövergripande CRISPR‑verktyg. De konstruerade humana fibroblaster så att IL‑11‑produktion kunde övervakas med en fluorescerande tagg, och utförde sedan parallella skärmar som antingen slog ut gener eller aktiverade dem över genomet. Genom att sortera celler som gjorde ovanligt höga eller låga mängder IL‑11 efter TGFβ‑ och IL‑1β‑stimulering identifierade de gener som kontrollerar detta svar. Bland många signaleringskomponenter framträdde en transkriptionsfaktor—GLIS3—som en huvudregulator. När GLIS3 inaktiverades producerade fibroblaster mycket mindre IL‑11; när den förstärktes steg IL‑11 kraftigt. Ytterligare experiment visade att GLIS3 flyttar in i fibroblastens kärna som svar på makrofagsignaler, binder direkt till DNA nära IL11‑genen och andra, och aktiverar ett brett program av inflammatoriska och fibrotiska gener, inklusive kollagener och faktorer som lockar fler immunceller. 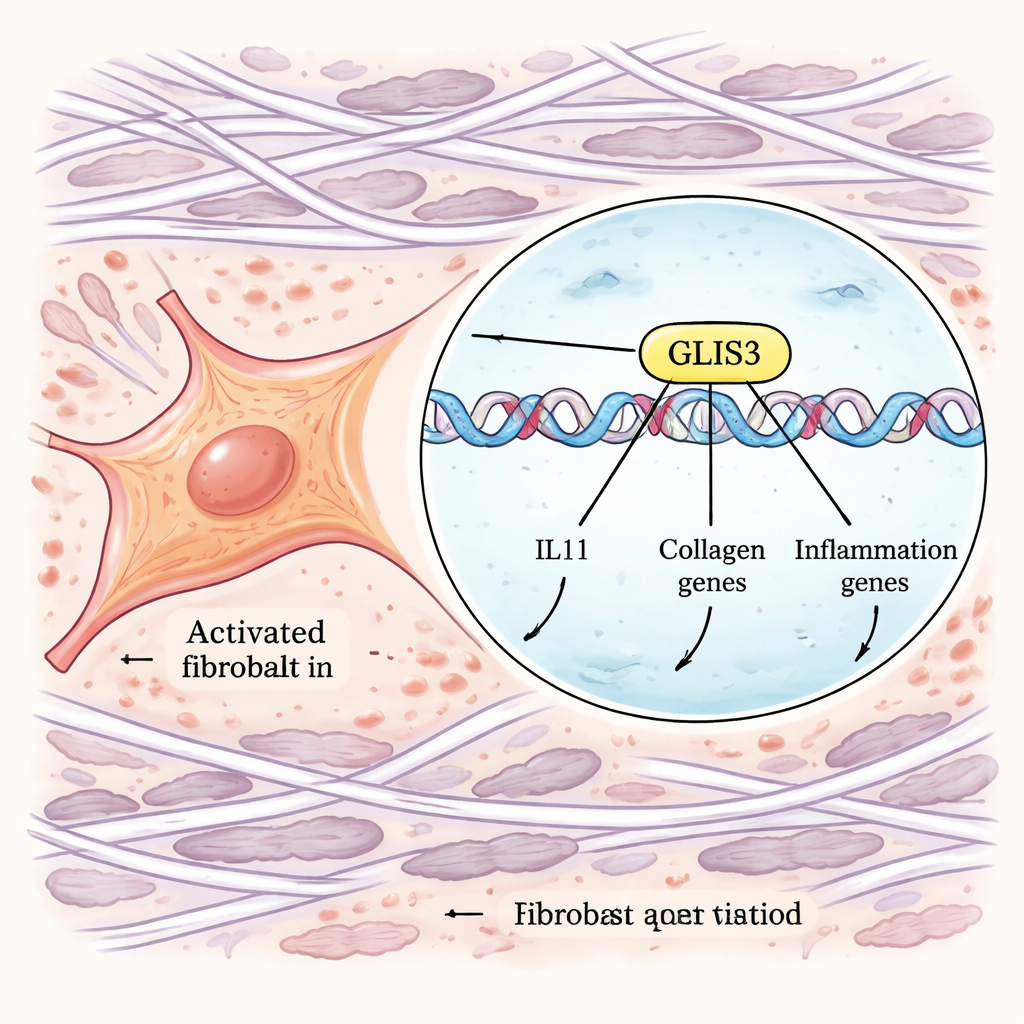
Från musmodeller till patienternas svårighetsgrad
Teamet frågade sedan om detta GLIS3‑drivna program spelar roll i levande organismer. Hos möss skapade de en stam där GLIS3 endast kunde tas bort från fibroblaster. När dessa djur utsattes för kronisk kolit utvecklade de mindre tarmärrbildning, hade lägre nivåer av kollagen och uttryck av fibrotiska gener, och visade minskad inflammation jämfört med normala möss. Rumslig kartläggning bekräftade att GLIS3‑defekta möss hade färre IL‑11‑producerande fibroblaster och färre närliggande aktiverade makrofager och neutrofiler, vilket indikerar att störning av GLIS3 försvagar hela den inflammatoriskt‑fibrotiska kretsen. Genom att vända sig till en stor pediatrisk kohort med ulcerös kolit destillerade författarna en GLIS3‑”signatur” på 50 gener och fann att dess aktivitet i colonbiopsier nära korrelerade med sjukdomens svårighetsgrad och förekomsten av IAFs och aktiverade makrofager, vilket kopplar denna väg direkt till patientutfall.
Att bryta cykeln av inflammation och ärrbildning
För icke‑specialister är slutsatsen att detta arbete avslöjar en självförstärkande loop: inflammatoriska makrofager triggar fibroblaster att bli ärrbildande IAFs; dessa IAFs, under kontroll av GLIS3, pumpar ut IL‑11, kollagen och andra faktorer som omformar vävnaden och lockar fler inflammatoriska celler. Vanliga läkemedel som brett dämpar immunsystemet kanske inte helt bryter denna loop, vilket bidrar till att förklara varför många patienter så småningom misslyckas med befintliga behandlingar. Genom att identifiera GLIS3 och det IL‑11‑producerande fibroblasttillståndet som centrala noder i inflammations‑fibros‑kretsen pekar denna studie mot mer riktade strategier—inriktade på fibroblaster snarare än enbart immunceller—som en dag kan förhindra eller vända ärrbildning vid inflammatorisk tarmsjukdom och möjligen andra kroniska inflammatoriska tillstånd.
Citering: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Nyckelord: inflammatorisk tarmsjukdom, intestinal fibros, fibroblaster, makrofager, GLIS3