Clear Sky Science · sv
Effektiv och noggrann rumslig sammanblandning av maskinlärda interatomära potentialer för materialvetenskap
Varför snabbare atomära simuleringar spelar roll
Att ta fram bättre material för tekniker som fusionsenergi, mikroelektronik och konstruktionslegeringar förlitar sig i allt större utsträckning på datorbaserade simuleringar som följer hur atomer rör sig och interagerar. De mest exakta metoderna bygger på kvantfysikens principer, men de är så beräkningsintensiva att bara måttliga systemstorlekar och tidsskalor är praktiskt genomförbara. Denna artikel introducerar ML‑MIX, en teknik och programvara som låter forskare behålla närapå kvantmekanisk noggrannhet precis där den behövs, samtidigt som enklare och billigare modeller används i övrigt. Resultatet är en väsentlig hastighetsökning—ofta en faktor 4 till 10—utan att frångå tillförlitligheten i de centrala fysikaliska förutsägelserna.
Att blanda detaljerade och enkla bilder av atomer
Kärnan i arbetet är en enkel idé: inte varje atom i en simulering behöver samma nivå av detaljer. Regioner där bindningar töjs, bryts eller omorganiseras—såsom defekter, ytor eller implanterade partiklar—drar nytta av moderna maskinlärda interatomära potentialer, vilka efterliknar kvantmekanisk noggrannhet. Men atomer långt från dessa ”hetpunkter” vibrerar mest omkring regelbundna positioner och kan hanteras av betydligt enklare modeller. ML‑MIX ger ett sätt att kombinera en exakt men dyr modell med en slankare, ”billigare” modell i samma simuleringslåda. Det görs genom att definiera en kärnzon som använder den dyra modellen, en omgivande buffert där krafterna noggrant blandas mellan modellerna, och en yttre bulkzon som endast använder den billiga beskrivningen. 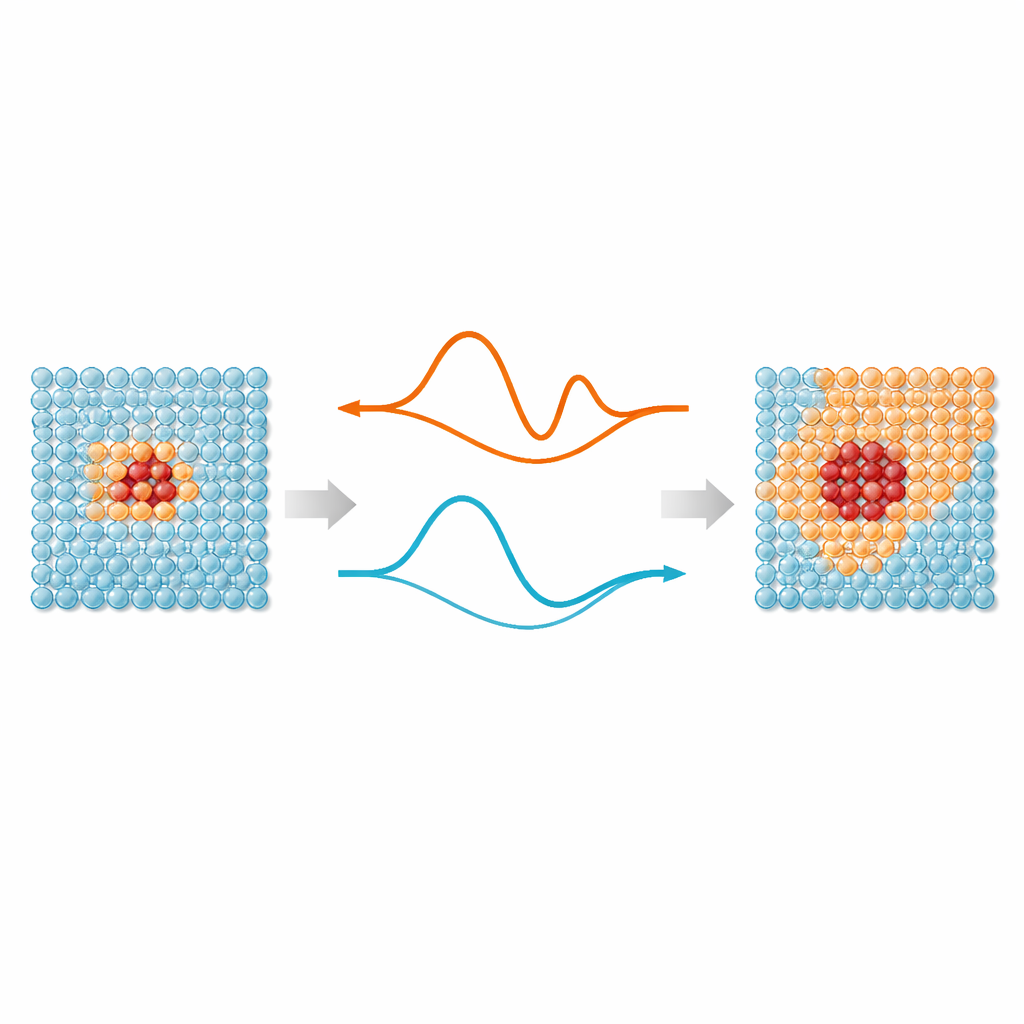
Att lära en billig modell att efterlikna en exakt
En viktig utmaning är att försäkra att den billiga modellen beter sig som den exakta där de möts. Istället för att passa den billiga modellen direkt mot en omfattande och varierad kvantmekanisk datamängd genererar författarna fokuserade ”syntetiska” data genom att köra den exakta modellen under de specifika förhållanden som är relevanta för bulkregionen: högtemperaturvibrationer och lätt påspända kristaller. De passar sedan den billiga modellen så att den matchar dessa data, samtidigt som strikta begränsningar införs för grundläggande materialegenskaper som elastiska konstanter och gitteravstånd. Denna begränsade anpassning säkerställer att långräckviddiga spänningar och deformationer överensstämmer smidigt över gränsen mellan de två modellerna, och undviker artificiella krafter som annars skulle kunna fördärva dynamiken vid gränsytan.
Metodens prövning
För att kontrollera att ML‑MIX verkligen fungerar kör författarna en rad tester på system av kisel, järn och volfram. För ett enkelt exempel beräknar de energibarriären för en vakans—en tom gitterplats—i kisel att förflytta sig från en position till en annan. Den blandade simuleringen återger resultatet från en helt dyr beräkning inom en tusendels elektronvolt, samtidigt som den går ungefär fem gånger snabbare. I en mer dynamisk situation töjer de en enskild kiselbindning i en het kristall och mäter den genomsnittliga kraften på den. En simulering som använder endast den billiga modellen kommer redan förvånansvärt nära, men när en liten dyr kärna läggs till runt den utdragna bindningen blir överensstämmelsen statistiskt omöjlig att skilja från den fullt exakta referensen, med hastighetsökningar på upp till ungefär 13 gånger i seriekörningar.
Följa defekter och partiklar i rörelse
Mer realistiska tester undersöker hur defekter rör sig genom metaller. Teamet simulerar diffusionen av en självinterstitiell defekt i järn och av helium‑atomer i volfram. I varje fall begränsas den dyra modellen till en liten rörlig region runt defekten, medan resten av kristallen hanteras av den billiga potentialen. De resulterande diffusionskoefficienterna överensstämmer med de från fullt exakta simuleringar inom statistisk felmarginal, även när en enbart billig simulering skulle misslyckas. Författarna pressar sedan metoden till större, vetenskapligt viktiga problem i volfram, ett ledande kandidatmaterial för fusionsreaktorer. De modellerar rörelsen hos skruvdislokationer—linjeliknande defekter som styr plastisk deformation—och implantationen av helium‑atomer i en het volframyta. I båda fallen återger ML‑MIX resultaten från enbart den dyra modellen samtidigt som beräkningskostnaden minskas med faktorer på cirka fyra till elva. 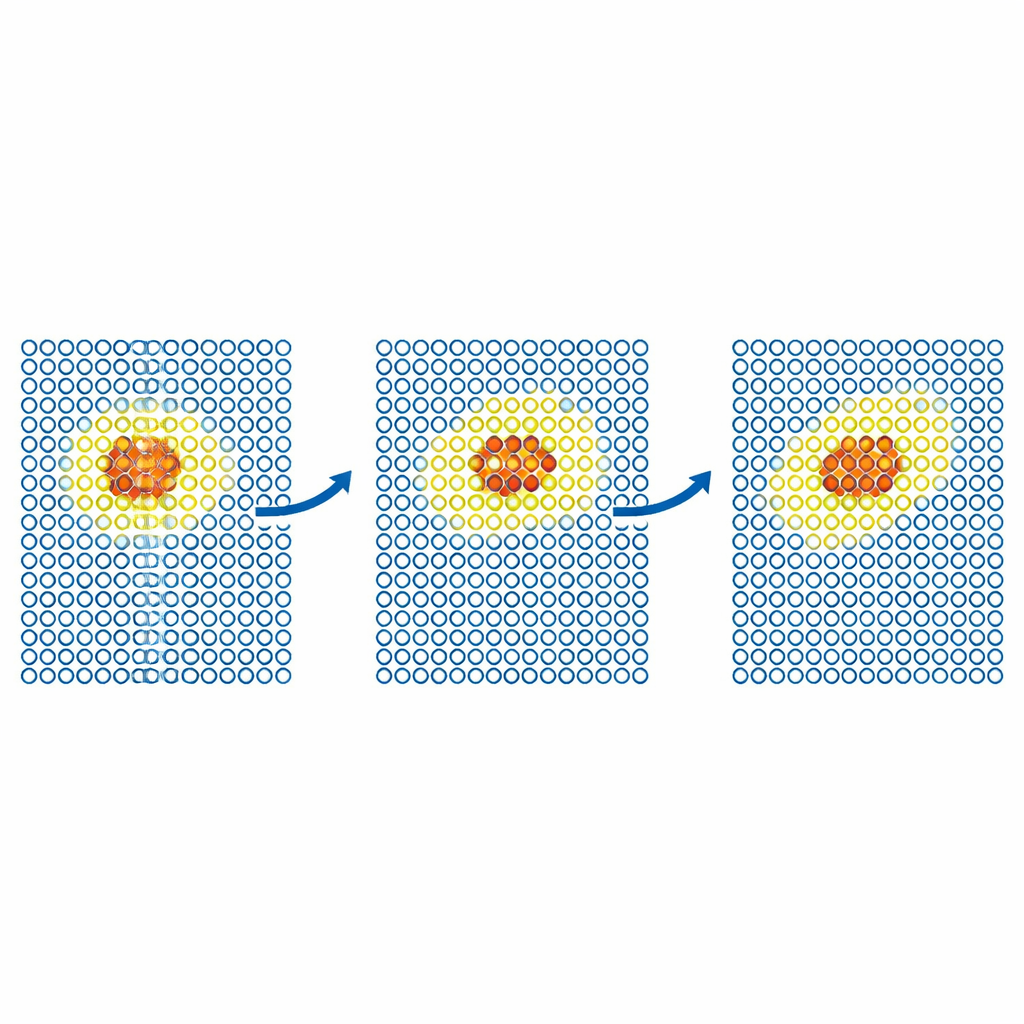
Jämförelse med experiment och framtidsutsikter
Studien av heliumimplantation visar tydligast styrkan i detta tillvägagångssätt. Genom att blanda en toppmodern maskinlärd modell för helium–volfram‑interaktioner med en snabbare potential för rent volfram simulerar författarna många fler kollisioner och större prover än vad som annars vore genomförbart, allt på grafiska processorer. Den beräknade andelen helium‑atomer som studsar av ytan jämfört med de som implanteras i metallen överensstämmer med experimentella mätningar upp till infallande energier runt 80 elektronvolt, något tidigare simuleringar hade svårt att uppnå. Även om blandningsschemat inte strikt bevarar energi och kräver milda termostater är den resulterande driften liten och hanterbar. Sammanfattningsvis visar ML‑MIX att noggrant sammansatta kombinationer av detaljrika och förenklade atommodeller kan överbrygga långvariga motsättningar mellan noggrannhet och skala, och öppna dörren för rutinmässiga, högfidelitetsimuleringar av komplexa material i realistiska miljöer.
Citering: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Nyckelord: maskinlärda interatomära potentialer, multiskalig materialsimulering, tungsten‑helium implantation, defekter och dislokationer, acceleration av molekylärdynamik