Clear Sky Science · sv
Grafisk atomklusterutvidgning för grundläggande maskininlärningsbaserade interatomära potentialer
Lära datorer att känna atomerna
Att utforma nya material för batterier, flygplan eller fusionsreaktorer handlar ofta om en enkel fråga: hur skjuter och drar atomer i varandra? Att beräkna dessa krafter exakt är så kostsamt att det kan ta dagar på en superdator för ett enda material. Denna artikel introducerar en ny familj maskininlärningsmodeller, kallad GRACE, som fungerar som en universell ”räknare” för atomära krafter över större delen av det periodiska systemet. Målet är att göra noggranna simuleringar av komplexa material rutinmässiga i stället för heroiska.
En enda modell för många material
De flesta befintliga maskininlärningsbaserade kraftfält är specialverktyg: de fungerar mycket bra för ett fåtal element eller föreningar, men måste byggas om från grunden när nya element läggs till. GRACE tar en annan väg. Den är utformad från början som en grundläggande modell som kan hantera 89 kemiska element och en enorm variation av atomära arrangemang med en gemensam uppsättning regler. För att uppnå detta bygger författarna vidare på ett matematiskt ramverk kallat atomic cluster expansion och utvidgar det till graf-liknande strukturer, vilket gör det möjligt för modellen att beskriva både lokala atomnärområden och mer utbredda mönster på ett enhetligt sätt. I stället för att hårdkoda varje möjlig interaktion lär sig GRACE kompakta ”inbäddningar” som fångar likheter mellan element, så att kunskap om ett material kan bidra till att beskriva ett annat.
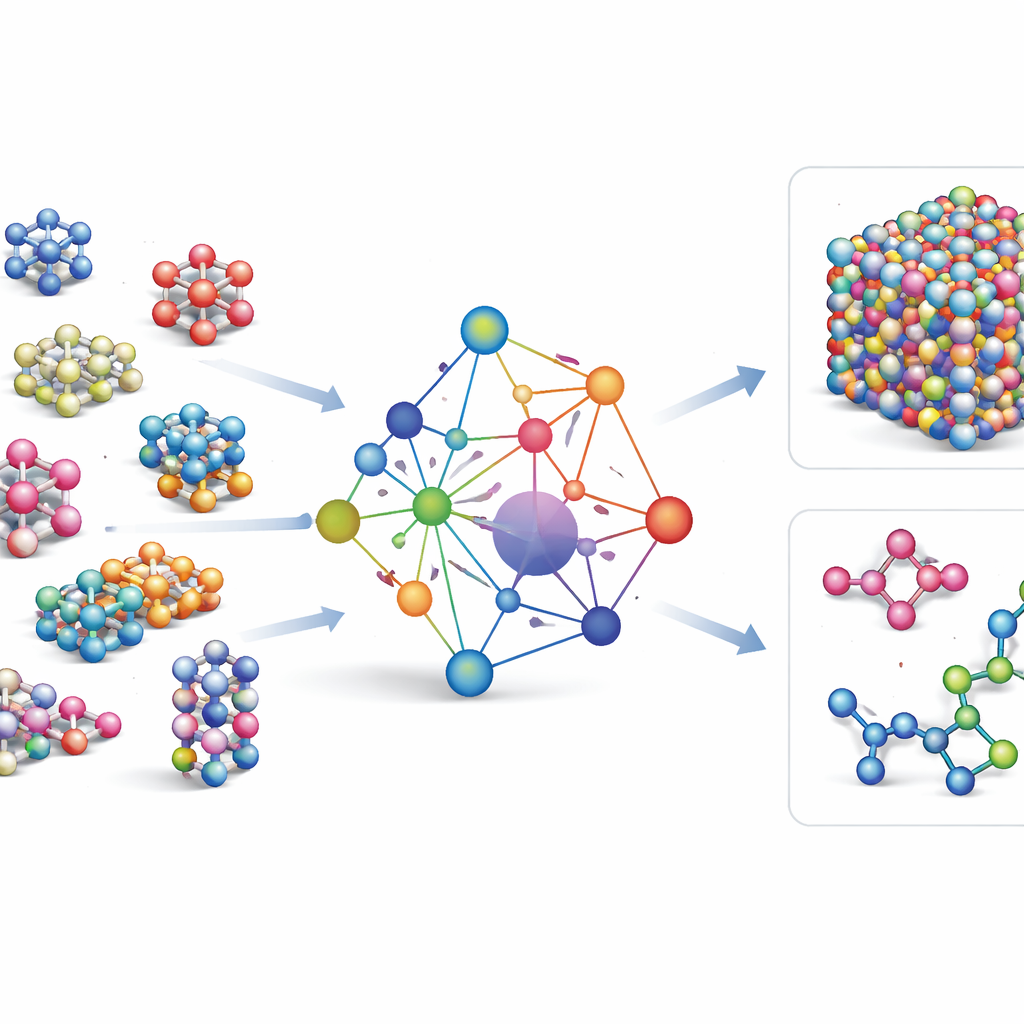
Träning på ett hav av atomdata
För att lära GRACE hur atomer beter sig samlade författarna några av de största publika databaserna med kvantmekaniska beräkningar. Kärnan är OMat24-samlingen, som innehåller omkring 110 miljoner simuleringar av oorganiska material, kompletterad med två andra som spårar hur strukturer relaxerar och utvecklas. Tillsammans täcker dessa datasätt nära-jämviktskristaller, påverkade och förvrängda strukturer, högtemperaturiga vätskor med mera, över samma breda urval av element. GRACE-modeller finns i flera storlekar, från enklare ett-lagersversioner som bara ser lokala atommiljöer till djupare två-lagersversioner som effektivt skickar ”meddelanden” mellan intilliggande regioner. Initial träning strävar efter en god balans mellan energier, krafter och interna spänningar, och vidare finslipning justerar modellerna för att vara kompatibla med allmänt använda referensdatabaser inom materialvetenskapen.
Sätta modellen på prov
En universell modell är bara användbar om den presterar tillförlitligt över många uppgifter. Författarna utsätter därför GRACE för ett krävande testpaket som speglar hur forskare faktiskt använder atomistiska simuleringar. På ett samhällsbenchmark för att upptäcka stabila kristallstrukturer ligger GRACE konsekvent på ”Pareto-fronten”: för en given noggrannhet är den snabbare än konkurrerande modeller, och för en given hastighet är den mer exakt. Liknande fördelar syns vid prediktion av termisk ledningsförmåga, en egenskap som är känslig för mycket små förändringar i atomrörelser. GRACE presterar också väl vad gäller elastiska egenskaper, ytenergi, korngränsenergier och punktdefektbildningsenergier i många rena metaller, vilka alla prövar hur material reagerar på att bli utsträckta, skurna eller lokalt skadade. Ett långt molekylärdynamiklopp av ett hett smält salt visar att modellen förblir numeriskt stabil i nanosekunder samtidigt som den återger detaljerade strukturmönster och atomdiffusionshastigheter.
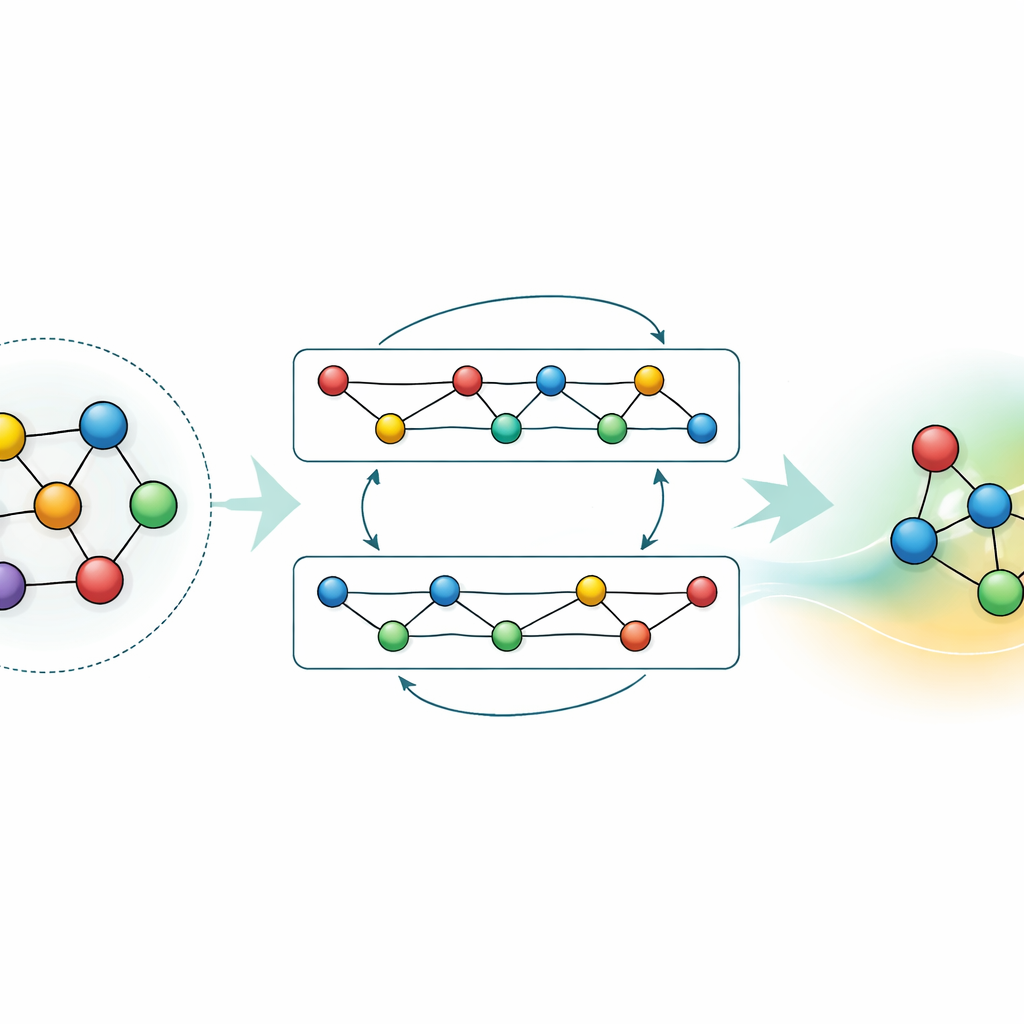
Anpassa och komprimera kunskapen
Medan en allmän modell är kraftfull kräver många tillämpningar antingen högre noggrannhet för ett specifikt material eller snabbare beräkningar på enklare hårdvara. Författarna visar två strategier för att uppnå detta utan att kasta bort vad GRACE redan lärt sig. Först finjusterar de den grundläggande modellen på fokuserade datasätt, såsom aluminium–litium-legeringar eller detaljerade väteförbränningsvägar. För legeringarna skärper även måttliga mängder extra data prediktionerna avsevärt och överträffar modeller som tränats från början med samma information. För förbränning skulle naiv finjustering normalt få modellen att ”glömma” vad den visste om andra material; genom att varsamt frysa delar av nätverket och bara uppdatera utvalda parametrar begränsar författarna detta katastrofala glömska samtidigt som de förbättrar noggrannheten för den nya kemin. För det andra visar de hur man destillerar den stora modellen till en mycket enklare ”student” som imiterar läraren på nyckelsystem. Denna destillerade version körs omkring sjuttio gånger snabbare på en CPU men bibehåller större delen av noggrannheten, särskilt när den tränats på en blandning av komplexa legeringar och enklare referensstrukturer märkt av den ursprungliga GRACE.
Vad detta betyder för framtida materialdesign
Arbetet positionerar GRACE som en flexibel grund för nästa generation av atomistisk modellering. I stället för att skapa en ny potential för varje material eller egenskap kan forskare utgå från en universell GRACE-modell och sedan finjustera eller destillera den efter sina behov, vilket sparar enorma mängder datorresurser och expertarbete. Benchmark-testerna visar att detta tillvägagångssätt inte bara matchar befintliga verktyg; det överträffar dem ofta både i hastighet och tillförlitlighet, särskilt för krävande egenskaper som värmetransport. För icke-specialister är huvudbudskapet att en enda, väl utformad maskininlärningsmodell nu kan fungera som en brett betrodd ”motor” för virtuella experiment över stora delar av det periodiska systemet, vilket påskyndar sökandet efter bättre batterier, katalysatorer, strukturella legeringar och energimaterial.
Citering: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Nyckelord: maskininlärningsbaserade interatomära potentialer, materialmodellering, atomära simuleringar, grundläggande modeller, grafisk atomklusterutvidgning