Clear Sky Science · sv
Ursaken till maskininlärningskraftfältens fel över metalliska grundämnen
Varför vissa metaller är svårare för AI att förstå
Maskininlärningsmodeller blir allt kraftfullare verktyg för att simulera hur atomer rör sig, och sparar forskare stora mängder beräkningstid jämfört med traditionella kvantberäkningar. Man skulle kunna förvänta sig att de enklaste materialen i naturen — rena metaller bestående av ett enda grundämne — vore de lättaste för dessa modeller att lära sig. Denna studie visar att så inte är fallet: vissa metaller förblir envist svåra att beskriva, och författarna avslöjar en fysisk förklaring till varför.
Att bygga en stor, ren karta över metalliskt beteende
För att systematiskt undersöka problemet skapade forskarna en ny datamängd kallad Metal-43, baserad på krävande kvantmekaniska beräkningar. Den täcker 43 olika metalliska grundämnen, från lätta litium till tunga volfram, alla behandlade med konsekventa beräkningsinställningar. För varje metall simulerade de tusentals atomstrukturer vid flera temperaturer och registrerade energi och krafter på varje atom. Denna noggrant kontrollerade ”lekplats” låter dem testa maskininlärningskraftfält — AI-modeller som förutsäger atomära krafter — under rättvisa och jämförbara förhållanden över många metaller. 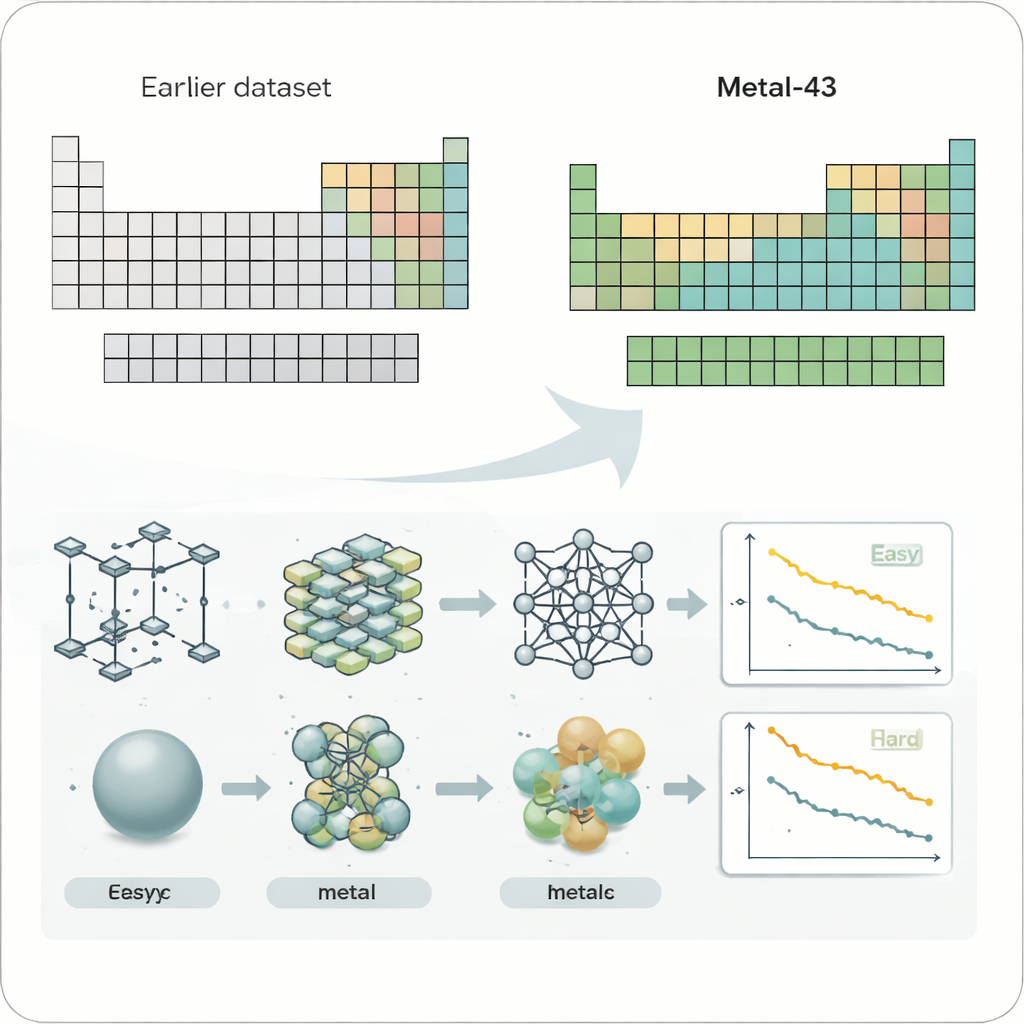
Hur modellfel följer periodiska systemet
Fyra vida använda maskininlärningskraftfältsmodeller undersöktes, inklusive både kompakta modeller tränade separat för varje grundämne och stora, allmänna modeller tränade på många system samtidigt. När författarna plottade prediktionsfelen i en periodiska systemets layout framträdde ett slående mönster. Mjuka, svagare bundna metaller som alkalimetaller och alkaliska jordartsmetaller tenderade att vara enklare för alla modeller, medan tidiga övergångsmetaller i mitten av tabellen — de som ofta används i högpresterande legeringar och katalysatorer — konsekvent gav mycket större fel. Denna trend höll i sig även när de råa felen omskalades för att ta hänsyn till den övergripande styrkan i atomkrafterna, vilket visar att svårigheten inte bara handlar om starkare bindningar utan något mer grundläggande.
Gömd komplexitet i metallens elektroniska ”trafikkarta”
Arbetets nyckelinsikt är att koppla dessa modellfel till formen på varje metals Fermiyta, vilket är en slags tredimensionell ”trafikkarta” över var elektroner kan röra sig vid de energier som är viktigast. I lättanpassade metaller är denna yta slät och nära sfärisk. I svåranpassade tidiga övergångsmetaller blir den taggig och ficklik, vilket speglar komplicerat elektroniskt beteende kopplat till delvis fyllda d-orbitaler. När atomer skjuts till eller förskjuts något förändras dessa intrikata Fermiytor på ett ojämnt, ibland abrupt sätt, vilket i sin tur gör den totala energilandskapet skrovligt och komplext. Författarna visar att enkla numeriska mått på hur snabbt vissa elektroniska energisummor svänger vid små störningar korrelerar starkt med hur stora maskininlärningsfelen är, särskilt för de problematiska övergångsmetallerna. 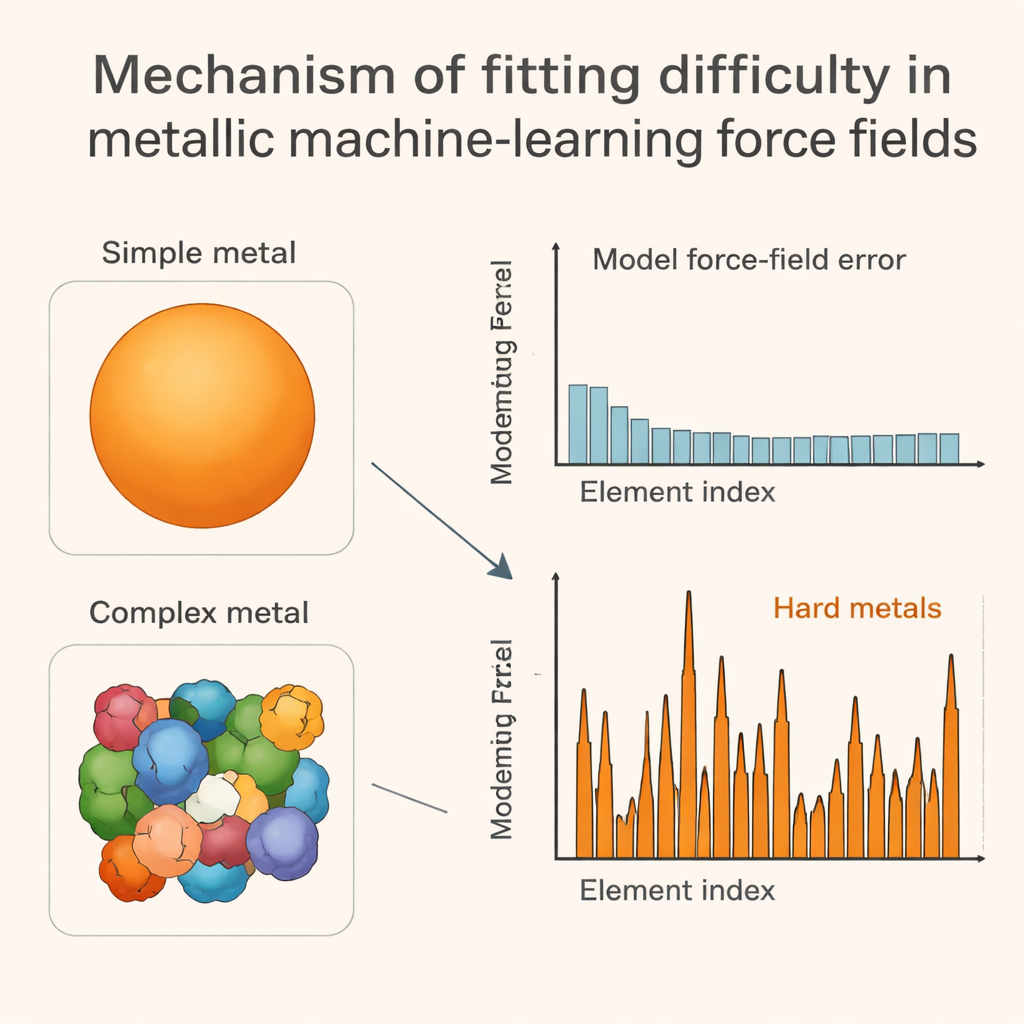
Begränsningar i dagens AI-modeller, även på idealiserade data
För att skilja på metallernas egen svårighet och begränsningarna i dagens AI-ansatser genererade teamet också artificiella datamängder med traditionella, handgjorda modeller för atomära krafter. Vissa av dessa äldre modeller beror huvudsakligen på avstånd mellan atomer, medan andra inkluderar stark vinkelberoende som efterliknar mer riktad bindning. Maskininlärningskraftfält kunde reproducera de avståndsbaserade modellerna nästan perfekt, men deras fel ökade kraftigt när angleffekter var viktiga — särskilt för metaller som redan var kända som svåra. Denna jämförelse visar att utmaningen inte bara ligger i metallernas underliggande fysik utan också i representerbarheten hos dagens maskininlärningsarkitekturer, som fortfarande har svårt med starkt vinkelberoende, mångkroppsinteraktioner.
Vad detta betyder för framtida simuleringar
För icke-specialister är huvudslutsatsen att det finns en tydlig, fysiskt grundad förklaring till varför vissa metaller är mycket svårare för AI att modellera än andra: komplexiteten i hur deras elektroner rör sig vid Fermiytan gör energilandskapet skrovligt och invecklat. Metal-43-datamängden och de enkla indikatorerna från elektronstruktur som föreslås här ger forskare ett sätt att förutse vilka material som blir problematiska, att benchmarka nya modeller rättvist och att utforma förbättrade kraftfält som bättre fångar riktad bindning. På längre sikt bör dessa insikter bidra till att göra AI-baserade simuleringar mer pålitliga för design av avancerade legeringar, katalysatorer och annan metallbaserad teknik.
Citering: Geng, X., Zhang, W., Wang, LW. et al. Origin of the machine learning forces field errors across metal elements. npj Comput Mater 12, 102 (2026). https://doi.org/10.1038/s41524-026-01977-3
Nyckelord: maskininlärningskraftfält, metalliska material, Fermiyta, interatomära potentialer, densitetsfunktionalteori