Clear Sky Science · sv
Självoptimerande maskininlärningspotential‑stödd automatiserad arbetsflöde för mycket effektiv materialsdesign av komplexa system
Smartare sökningar efter nya material
Att ta fram nya material liknar att leta efter en nål i en nästintill oändlig höstack. Från bättre batterier och snabbare datorer till effektivare lasrar och potentiella rumstemperatursuperledare—många framtida teknologier vilar på att man hittar precis rätt atomarrangemang. Denna artikel presenterar ett sätt att låta artificiell intelligens sköta större delen av sökandet automatiskt, vilket dramatiskt minskar tid och kostnad för att hitta lovande nya föreningar.
Varför materialpusslet är så svårt
Ett solids egenskaper—hur väl det leder elektricitet, hur starkt det är, hur det reagerar på ljus—bestäms av hur dess atomer är ordnade i tredimensionella mönster som kallas kristallstrukturer. I teorin kan man använda kvantmekanik för att beräkna vilka arrangemang som är stabila och vilka egenskaper de får. I praktiken är dessa kvantberäkningar så krävande att endast en liten bråkdel av alla möjliga material kan kontrolleras. Utmaningen växer snabbt när fler än två grundämnen är inblandade, eftersom antalet kombinationer och atomära arrangemang exploderar och gör en blint sökning omöjlig.
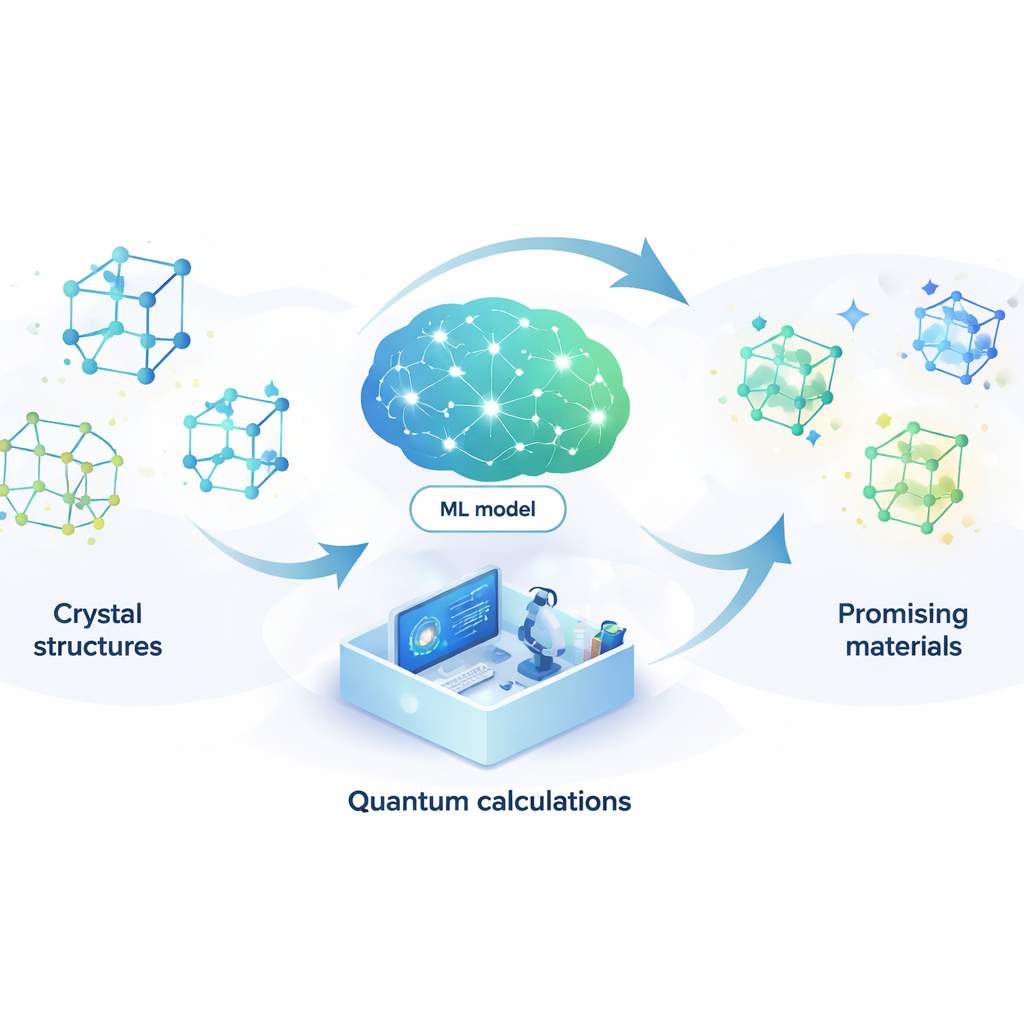
Låta en inlärningsmodell ersätta kvantfysiken
För att angripa problemet bygger författarna en maskininlärningsmodell som kan efterlikna resultaten från kostsamma kvantberäkningar till en bråkdel av kostnaden. Deras modell, kallad attention-coupled neural network (ACNN), lär sig hur ett materials energi beror på atomernas positioner och typer. När den väl är tränad kan den mycket snabbt uppskatta om en föreslagen kristallstruktur sannolikt är stabil eller inte, och vilka krafter som verkar på varje atom. Viktigt är att modellen är utformad för att respektera grundläggande fysikaliska krav, såsom att förskjutning eller rotation av hela kristallen inte ska ändra dess totala energi.
En självförbättrande upptäcktsloop för material
I stället för att träna modellen en gång och hoppas att den fungerar överallt, kapslar författarna in den i en självoptimerande loop. Processen börjar med en liten uppsättning slumpmässiga kristallstrukturer som utvärderas med fullständiga kvantmekaniska beräkningar och används för att träna en initial ACNN. Denna modell används sedan för att relaxera miljontals provstrukturer och hittar snabbt lokala energiminima—kandidater för stabila eller nära stabila faser. Arbetsflödet flaggar automatiskt två särskilt värdefulla typer av strukturer: de som ser mycket stabila ut och de som framstår som ofysiska eller misstänkta. Endast dessa utvalda fall skickas tillbaka till den kostsamma kvantlösaren, och de nya resultaten matas in i modellen för reträning. Över många omgångar blir modellen stadigt mer exakt i de delar av strukturutrymmet som är mest relevanta.
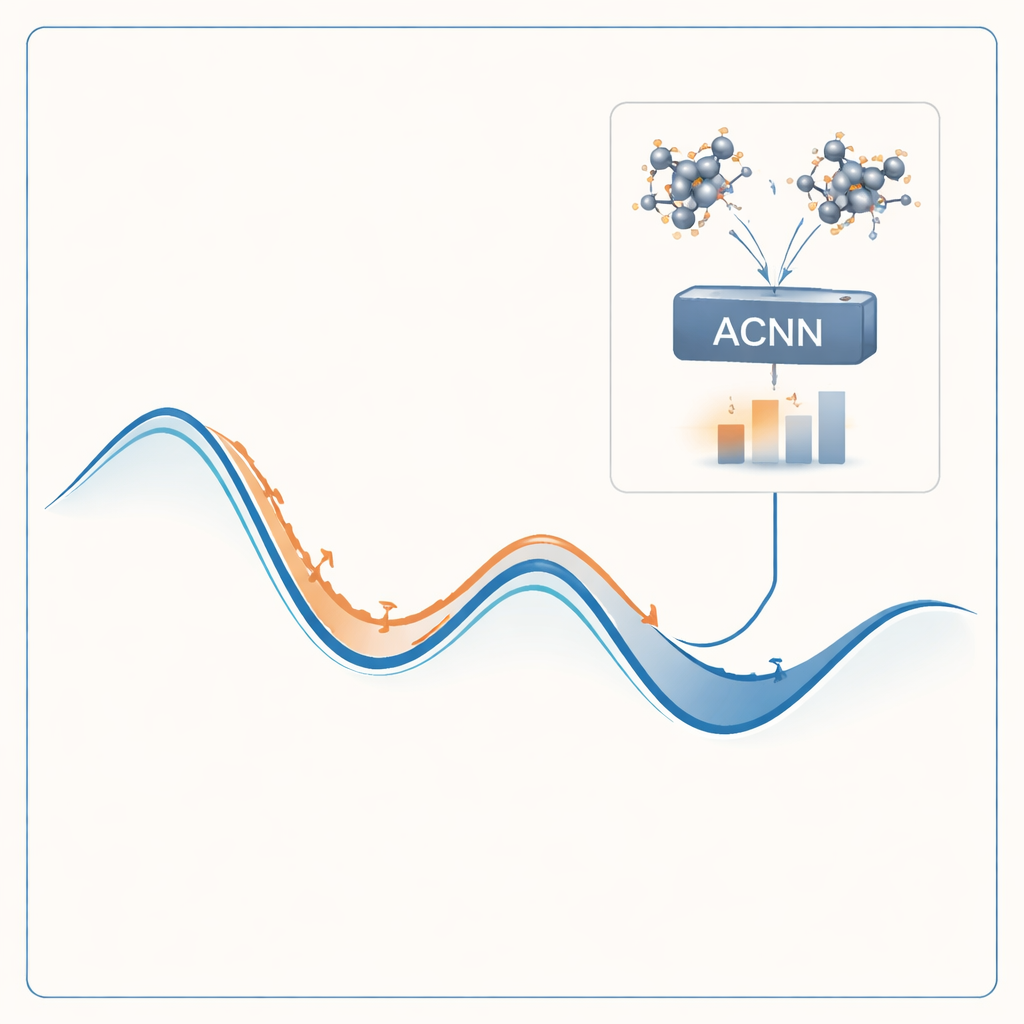
Metodens prövning
Gruppen demonstrerar sitt tillvägagångssätt på två krävande system. Det första är en högtrycksblandning av magnesium, kalcium och väte — en familj föreningar av stort intresse för högtemperatursuperledningsförmåga. Genom att utforska nära sex miljoner provstrukturer upptäcker deras arbetsflöde en ny stabil fas, MgCa₃H₂₃, och flera närbesläktade väterika "bur"-strukturer. Beräkningar tyder på att några av dessa kan bli superledande vid temperaturer över flytande kväves kokpunkt under extremt tryck. Det andra testet riktar sig mot ett fyrkomponentsystem innehållande beryllium, fosfor, kväve och syre, valt för sin potential att hysa kristaller som effektivt omvandlar laserljus till djup ultraviolett. Här relaxerar metoden mer än nio miljoner strukturer och identifierar tre termodynamiskt stabila faser med mycket breda bandgap och lovande optiska egenskaper.
Från brutal kraft till vägledd upptäckt
I båda exemplen uppnår det automatiserade arbetsflödet hastighetsökningar på ungefär tio tusen gånger jämfört med att enbart använda kvantberäkningar, samtidigt som det fortfarande pålitligt identifierar strukturer värda vidare studier. För en icke‑specialist är huvudbudskapet att en stor del av materialupptäckten nu kan hanteras av ett inlärningssystem som lär sig var det är osäkert och endast begär riktade, högprecisionsberäkningar när det behövs. Denna typ av självkorrigerande, AI‑stödda sökning öppnar dörren för att utforska betydligt mer komplexa elementblandningar än tidigare möjligt, och ökar chanserna att hitta nya superledare, optiska kristaller och andra funktionella material som ligger till grund för nästa generations teknologier.
Citering: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Nyckelord: materials discovery, machine learning potentials, crystal structure prediction, superconducting hydrides, nonlinear optical crystals