Clear Sky Science · sv
Lära sig dataeffektiv grovskalig molekylär dynamik från krafter och brus
Varför det spelar roll att krympa molekyler
Att simulera varje atoms rastlösa rörelse i ett protein och dess omgivande vatten är ett av våra bästa verktyg för att förstå hur livet fungerar på molekylär nivå. Men dessa all-atom-simuleringar är så beräkningsintensiva att det kan ta månader på en superdator att följa ett protein när det viks, veckas upp eller interagerar med andra molekyler under biologiskt relevanta tider. Denna artikel presenterar ett nytt sätt att bygga snabba, förenklade modeller av proteiner som ändå beter sig som sina fulla atomära motsvarigheter, samtidigt som de kräver avsevärt mindre träningsdata och beräkningsresurser än tidigare.
Från varje atom till en enklare bild
Traditionell molekylär dynamik spårar varje atom och beräknar krafterna mellan dem vid varje liten tidssteg. För att snabba upp beräkningarna använder forskare ofta grovskaliga modeller, som grupperar många atomer till ett mindre antal ”pärlor”. Dessa reducerade modeller körs mycket snabbare men har historiskt haft svårt att nå samma noggrannhet som fulla atomistiska simuleringar, särskilt för proteiner med rik fällningsdynamik. Nyare arbete har vänt sig till maskininlärning för att automatiskt upptäcka bättre grovskaliga kraftfält, men träning av dessa modeller har vanligen krävt miljontals detaljerade ögonblicksbilder, vardera märkta med krafterna på varje atom—en enorm datamängd och beräkningsbörda.
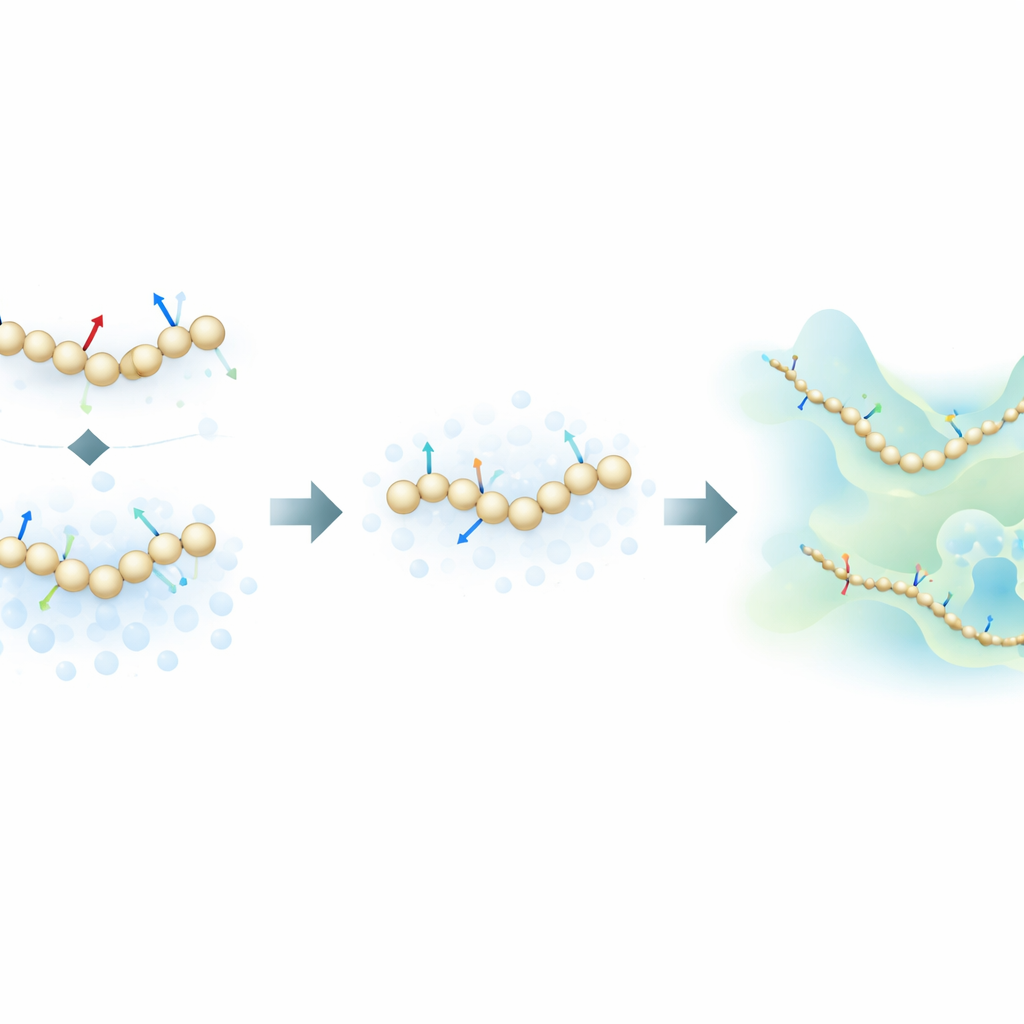
Att blanda fysiska krafter med informativt brus
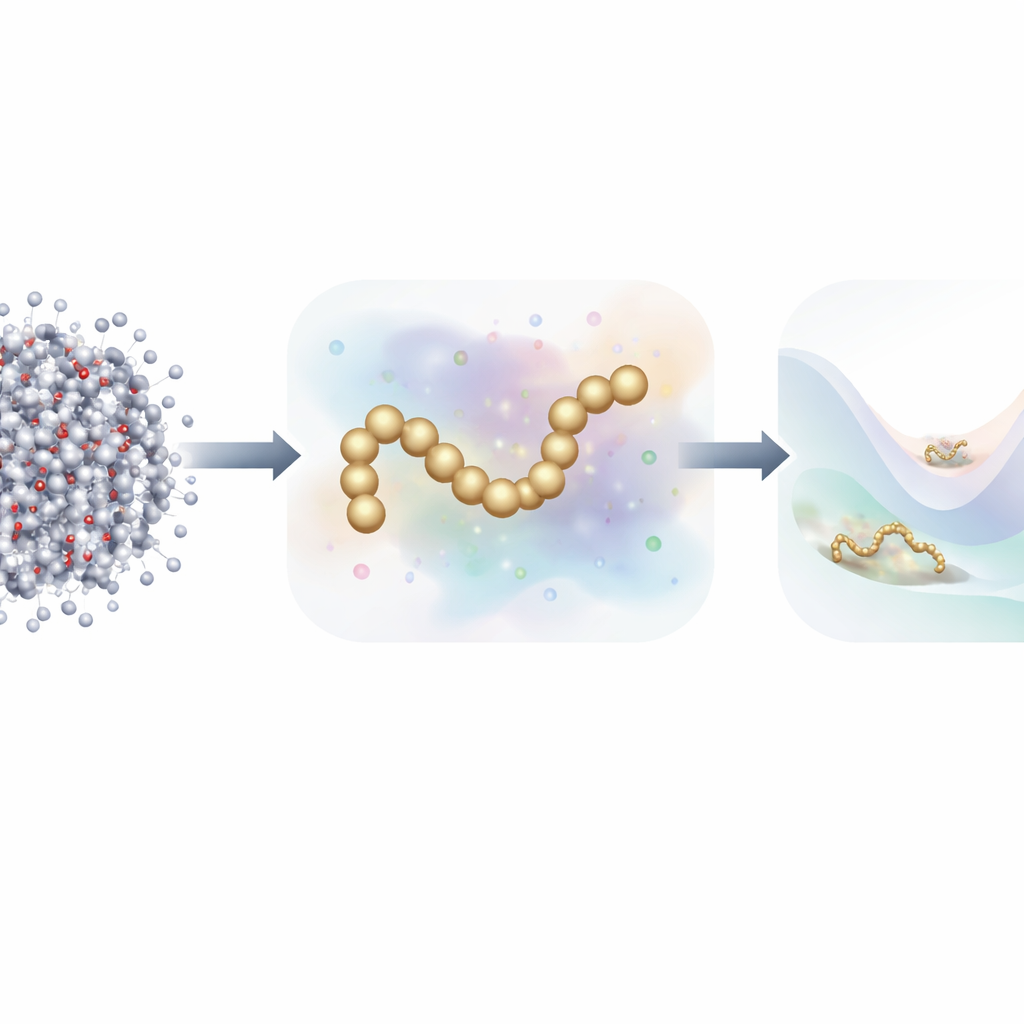
Författarna föreslår en ny träningsstrategi som hämtar inspiration från generativa diffusionsmodeller—samma klass av algoritmer som ligger bakom många moderna AI-bildgeneratorer. Istället för att enbart lära från de fysiska krafterna beräknade i atomistiska simuleringar, lär sig deras metod också från hur molekylstrukturer fördelar sig i rummet genom att avsiktligt tillsätta kontrollerat brus till grovskaliga konfigurationer. I denna ram är brus inte bara en störning som måste tas bort; det blir en extra informationskälla. Genom att matematisk förena den traditionella ”force matching”-ansatsen med avbrusningstekniker från diffusionsmodeller kan metoden sluta sig till det underliggande energilandskapet för ett protein med långt färre märkta exempel.
Lära enkla modeller att efterlikna komplexa proteiner
För att testa idén tränade forskarna neurala nätverksbaserade grovskaliga modeller för flera proteiner med ökande komplexitet: de små miniproteinerna Chignolin och Trp-Cage, det något större NTL9, och det 76-residiga proteinet Ubiquitin. De jämförde tre träningslägen: endast atomistiska krafter, endast brusbasserad information, och en kombination av båda. För de mindre proteinerna visade den nya kombinerade ansatsen att den kan återge huvuddragen i fällningslandskapet—såsom relativ stabilitet mellan vikt och ovikt tillstånd och förekomst av intermediärer—med upp till hundra gånger mindre träningsdata än standard force-matching-metoder. Överraskande nog matchade eller överträffade i datafattiga regime ofta även modeller tränade enbart med brusbasserad information noggrannheten hos kraftbaserad träning.
Att nå större och tuffare proteinsystem
Ubiquitin är ett mer krävande test: att fånga dess fällning och uppvikning vid realistiska temperaturer har historiskt krävt specialiserad hårdvara och extremt långa atomistiska körningar. Här tränar författarna grovskaliga modeller med en modest datamängd bestående av korta jämviktsimuleringar runt det vikta tillståndet samt icke-jämvikts “utdragna” simuleringar som tvingar proteinet att sträckas. Trots detta snedvridna träningsset och avsaknaden av en perfekt atomistisk referens under samma förhållanden återger modellen tränad med både krafter och brus en realistisk bild där vikta och ovikta tillstånd samexisterar, med det vikta tillståndet som stabilitetsmässigt gynnat. I kontrast misslyckas en modell tränad endast på krafter med att stabilisera det vikta tillståndet alls, medan en endas brusmodell föredrar ovikta strukturer. Anmärkningsvärt är att ingen av de grovskaliga modellerna helt enkelt memorerar de extrema utsträckta formerna från träningsdatan, vilket tyder på att det inlärda energilandskapet är fysikaliskt meningsfullt och inte bara ett avtryck av indata-trajectorierna.
Vad detta betyder för framtida simuleringar
Genom att göra brus till en träningssignal och slå samman det med fysiska krafter visar detta arbete att exakta grovskaliga modeller av proteiner kan byggas från långt mindre och mindre ideala datamängder än man tidigare trott. I praktiken innebär det att forskare kanske inte längre behöver millisekundlånga atomistiska simuleringar på specialiserade superdatorer innan de kan utforska ett biomolekyls beteende med maskinlärda grovskaliga dynamiker. Istället kan mer blygsamma simuleringar på allmänt tillgänglig hårdvara vara tillräckliga för att träna kraftfulla reducerade modeller som fångar viktiga fällningsvägar och termodynamiska balanser. Även om frågor kvarstår om hur man bäst väljer och tolkar det tillsatta bruset och hur metoden kommer att prestera på ännu större, mer komplexa biomolekylära sammansättningar, sänker detta tillvägagångssätt avsevärt tröskeln för att använda data-drivna grovskaliga simuleringar som ett rutinverktyg i molekylär vetenskap.
Citering: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Nyckelord: grovskalig molekylär dynamik, maskininlärningskraftfält, proteinfällningssituationer, diffusionsmodeller i kemi, dataeffektiv simulering