Clear Sky Science · sv
En sjukdomsframkallande Tau‑mutation orsakar störningar i autofagi‑lysosom som begränsar Tau‑nedbrytning i en modell för frontotemporal demens
När hjärnans städlag halkar efter
Varför utvecklar vissa personer förödande minnes‑ och beteendeproblem årtionden före hög ålder? Denna studie tar sig an den frågan genom att zooma in på ett enda hjärnprotein, Tau, och de små cellulära ”återvinningscentraler” som normalt håller det under kontroll. Genom att observera levande mänskliga nervceller under extremt skarpa mikroskop visar forskarna hur en sjukdomsframkallande Tau‑mutation täpper igen cellens avfallssystem och hur aktivering av detta system med en liten molekyl kan hjälpa till att rensa upp. Deras fynd kan peka mot nya behandlingsstrategier för vissa former av demens.
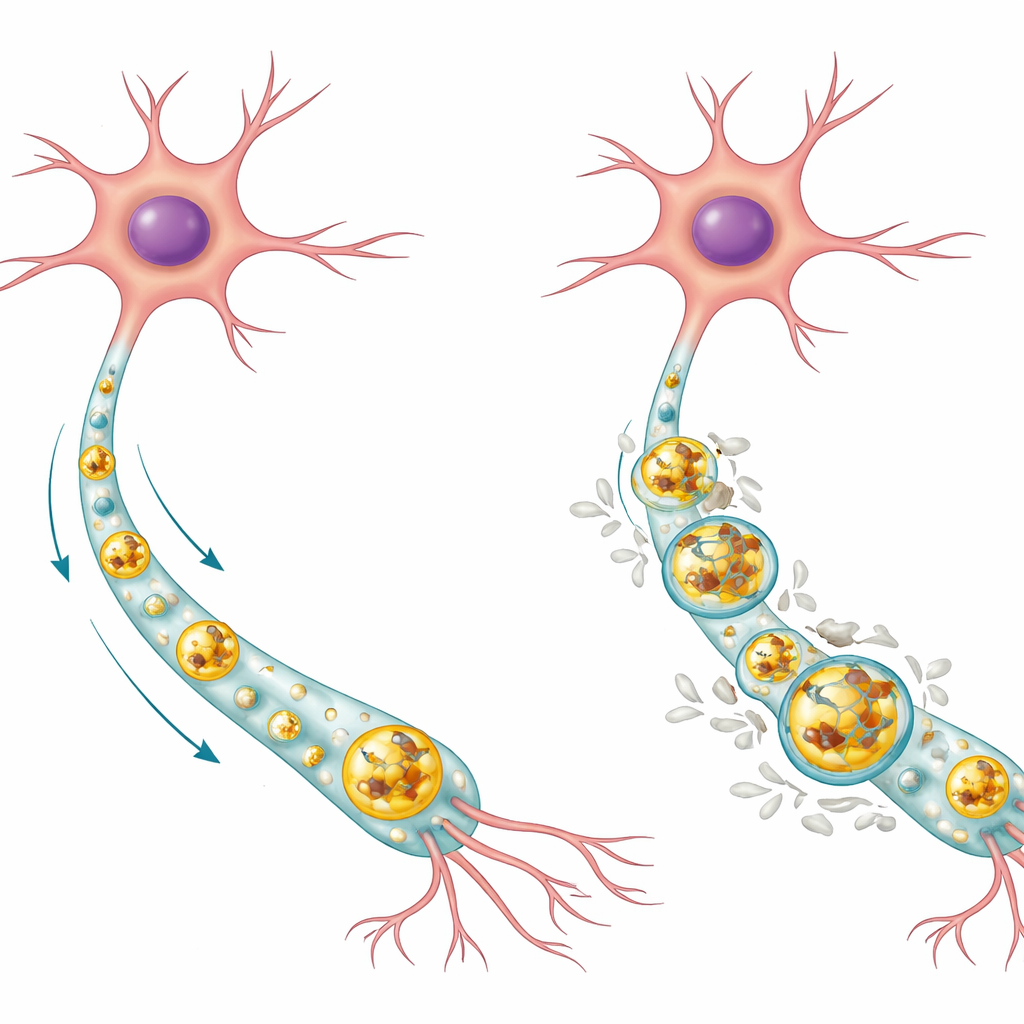
Hur hjärnceller vanligtvis tar ut soporna
Neuroner är långlivade celler som inte enkelt kan dela sig för att späda ut skadligt material, så de är starkt beroende av interna städsystem. En viktig väg är autofagi‑lysosom‑vägen. I denna process omsluts oönskade proteiner och utslitna delar i membranblåsor kallade autofagosomer, vilka sedan smälter samman med enzymfyllda kompartment kända som lysosomer, där lasten bryts ner och återvinns. I friska mänskliga neuroner fann författarna att normalt Tau tenderar att ackumuleras inne i lysosomens sura mitt, där det kan degraderas, medan fosforylerad Tau (en kemisk modifiering kopplad till sjukdom) i större utsträckning sitter på lysosomens yttre membran. De flesta lysosomer i friska celler var helt fria från Tau, vilket tyder på att detta system vanligtvis håller Tau‑nivåerna låga och välkontrollerade.
Vad som går fel i en genetisk form av demens
Teamet fokuserade på en mutation i MAPT‑genen, kallad p.R406W, som orsakar en ärftlig form av frontotemporal demens och kan efterlikna Alzheimer‑liknande minnesförlust. Med stamcellsteknik omprogrammerade de patienters hudceller till inducerade pluripotenta stamceller och vidare till stora mängder mänskliga neuroner som antingen bar mutationen eller hade genredigerats tillbaka till normal. I de mutanta neuronerna var total Tau och fosforylerad Tau markant högre, inte för att cellerna producerade mer Tau, utan för att de rensade det mindre effektivt. Superupplösningsavbildning visade att nästan alla lysosomer i de mutanta cellerna var fyllda med Tau och särskilt med fosforylerad Tau som belagde lysosommembranet. Denna ansamling signalerade att cellens huvudsakliga proteinborttagningsväg var blockerad.
Täppta återvinningscentraler och slö trafik
När de studerade återvinningsmaskineriet närmare såg forskarna att lysosomer i mutanta neuroner var fler, större och tenderade att ligga längre bort från cellkroppen. Live‑avbildning med fluorescerande färger visade att dessa lysosomer rörde sig långsammare och färdades kortare sträckor längs nervfibrerna, trots att de underliggande mikrotubuli‑spåren såg normala ut. De mutanta neuronerna innehöll också fler autofagosomer, mer av lasten‑adapterproteinet p62 och extra lipiddroppar — tecken på att material märktes för bortforsling men inte fullständigt bröts ner. Med en pH‑känslig rapportör fann de att autofagosomer i mutanta celler ofta misslyckades med att fusionera korrekt med lysosomer, vilket ledde till en ansamling av ”halvfärdiga” återvinningsvesiklar och breda fel i cellens städning, inte bara för Tau utan också för annat material.
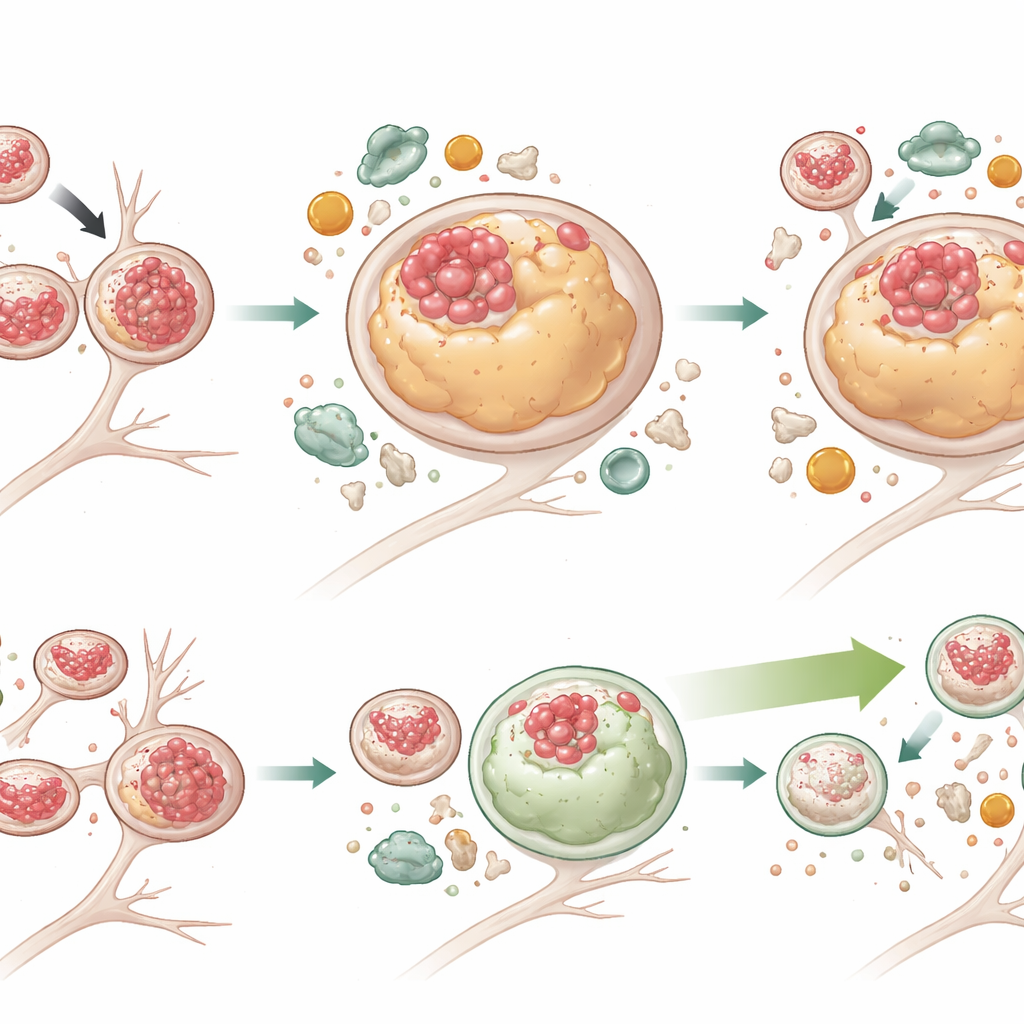
Stimulera cellens städning utan att lösa trafikstockningen
För att testa om förstärkning av autofagi kunde övervinna dessa problem behandlade teamet neuroner med G2‑567, en liten molekyl som tidigare visats stimulera autofagi‑lysosom‑systemet. Efter två veckors behandling hade mutanta neuroner avsevärt lägre nivåer av både total Tau och fosforylerad Tau, och många fler lysosomer var åter fria från Tau. Lysosomer krympte också tillbaka mot normal storlek. Markörer för aktiv autofagi ökade, medan p62 — en indikator på fastnad nedbrytning — föll i mutanta celler, vilket visar på mer effektiv nedbrytning av lasten. Intressant nog rättade inte G2‑567 alla defekter: lysosomer i mutanta neuroner tenderade fortfarande att ligga längre från cellkroppen och röra sig långsamt, och ett adapterprotein (JIP3) kopplat till lysosomtransport förblev förhöjt. Detta tyder på att lysosomers rörelse och deras nedbrytande funktion delvis kan separeras, och att förbättrad nedbrytning i sig kan räcka för att minska toxisk Tau‑ansamling.
Vad detta betyder för framtida demensbehandlingar
För en icke‑specialist är huvudpoängen att i denna genetiska modell av frontotemporal demens är problemet inte enbart att Tau blir onormal; det är att neuronernas återvinningssystem inte hinner med. p.R406W‑Tau‑mutationen stör direkt flera steg i autofagi‑lysosom‑vägen, vilket gör att Tau — särskilt dess fosforylerade form — ansamlas på och inne i lysosomer, tillsammans med annat icke‑nedbrutet material. Genom att farmakologiskt stimulera cellens städmaskineri kunde forskarna sänka Tau‑nivåerna och normalisera lysosomstorleken, även om transportdefekter bestod. Dessa resultat stärker idén att läkemedel utformade för att på ett säkert sätt öka autofagi och lysosomal funktion kan hjälpa återställa proteinbalansen vid tau‑relaterade demenser och kanske även vid vanligare tillstånd som Alzheimers sjukdom.
Citering: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Nyckelord: tauprotein, autofagi, lysosomdysfunktion, frontotemporal demens, neurodegeneration