Clear Sky Science · sv
DiNovo möjliggör hög täckning och hög tillförlitlighet vid de novo-peptidsekvensering via spegelproteaser och djupinlärning
Se proteiner i ny detalj
Proteiner är de små maskiner som håller våra celler vid liv, men att helt läsa deras byggstenar är fortfarande överraskande svårt. Denna artikel presenterar DiNovo, ett nytt mjukvarusystem som hjälper forskare att "läsa" proteindelar mycket mer komplett och tillförlitligt än tidigare. Genom att kombinera ett skickligt biokemiskt knep med modern artificiell intelligens lovar det att avslöja dolda proteiner, sjukdomsmarkörer och till och med immuntargets som traditionella metoder ofta missar.
Varför det är så svårt att läsa proteindelar
De flesta proteinanalyser i dag bygger på att man klyver proteiner i mindre bitar, kallade peptider, och sedan väger deras fragment i en masspektrometer. Från dessa massor försöker datorer återskapa den ursprungliga peptidsekvensen, ungefär som att lösa ett korsord utifrån ofullständiga ledtrådar. Befintliga metoder antar ofta att peptiderna kommer från kända proteindatabaser, vilket fungerar bra för välkända proteiner men har problem med nya eller oväntade sådana. Så kallad de novo-sekvensering undviker denna begränsning genom att försöka läsa peptider direkt från data, men den når ofta inte ända fram eftersom vissa fragment saknas och vissa peptider aldrig skärs rent från början.
Använda spegelenzym för att fylla luckorna
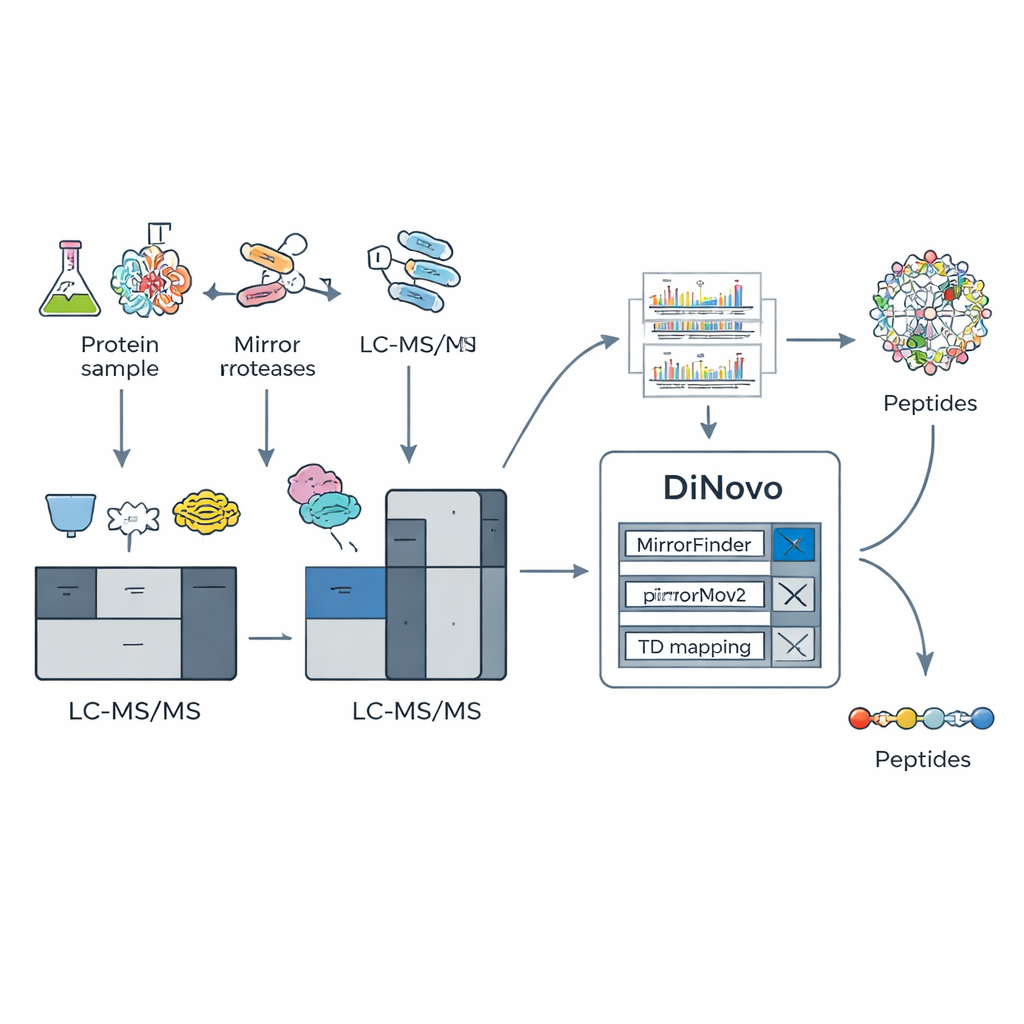
Huvudidén bakom DiNovo är att använda par av "spegelproteaser" – par av klyvningsenzym som skär proteiner på motsatta sidor av samma typ av aminosyra. Till exempel skär ett enzym precis före en lysin, medan dess partner skär precis efter den lysinen. Detta ger två besläktade peptider som delar samma inre segment men har olika ändar. När dessa "spegels"-peptider analyseras innehåller deras masspektra kompletterande fragmentmönster: vad som saknas i ett spektrum dyker ofta upp i det andra. Författarna visar att kombinationen av sådana spegelpar kan driva fragmenttäckningen nära komplett, med omkring 98 % av möjliga klyvningsställen stödda av verkliga experimentella signaler, betydligt högre än när man använder ett enda enzym.
En smart mjukvarupipeline byggd för spegeldata
För att utnyttja detta biokemiska trick byggde teamet DiNovo som ett helhetsverktyg. Först klyvs proteiner från bakterier och jäst med två spegelpar av enzymer, och de resulterande peptiderna analyseras med högupplöst masspektrometri. DiNovo använder sedan en modul kallad MirrorFinder för att automatiskt känna igen vilka par av spektra som kommer från spegelpeptider, och gör detta direkt från signalmönstren snarare än från några tidigare sekvensantaganden. Nästa steg, dess huvudsakliga de novo-motor MirrorNovo, använder djupinlärning för att tolka de parade spektrumen, medan en reservmotor baserad på grafmetoder, pNovoM2, erbjuder ett snabbare alternativ som bara kräver CPU. Tillsammans översätter dessa verktyg toppar till aminosyrasekvenser och granskar också de individuella spektrumen som inte bildade uppenbara par, för att pressa ut så mycket information som möjligt.
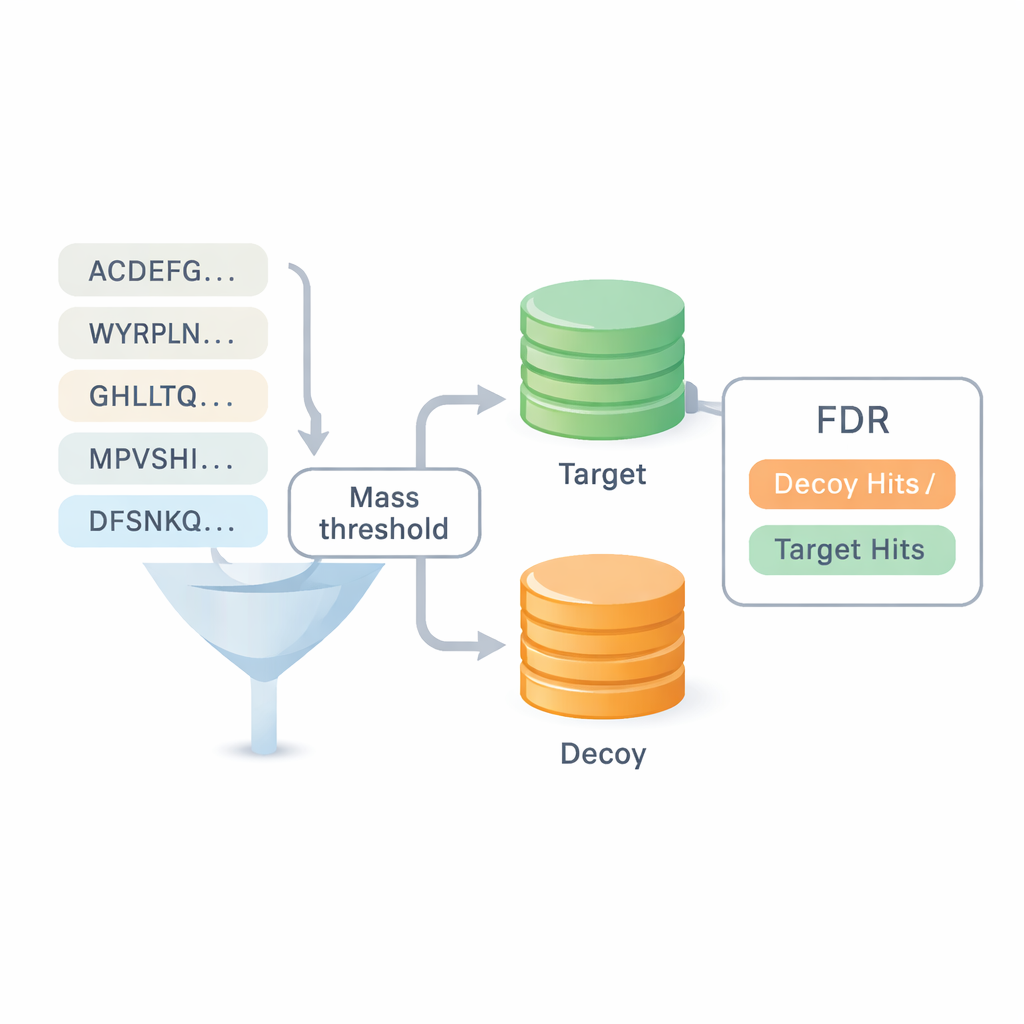
Mäta förtroende utan att förlita sig på gamla databaser
En av de största frågorna inom de novo-sekvensering är hur mycket man kan lita på resultaten. De flesta befintliga riktmärken återanvänder svar från databassökningar, vilket suddar ut gränsen mellan de två tillvägagångssätten och kan dölja fel. DiNovo introducerar en annorlunda kvalitetskontrollmetod kallad target-decoy-kartläggning. Här kartläggs de nyupplästa peptiderna mot en kombinerad samling av verkliga (target) och artificiella, slumpade (decoy) proteinsekvenser. Genom att jämföra hur ofta peptider hamnar i den verkliga uppsättningen kontra i den slumpade uppsättningen kan mjukvaran uppskatta en felprocent, eller false discovery rate, utan att förlita sig på tidigare identifieringar. Detta gör det möjligt att jämföra DiNovo direkt med standardprogram för databassökning under samma felkontroller.
Vad DiNovo levererar i praktiken
I tester på bakterie-, jäst- och antikroppsprover läste DiNovo konsekvent många fler peptider och aminosyror än välkända de novo-verktyg som använder endast ett enzym. Med två spegelpar producerade det 2–3 gånger fler högtillförlitliga aminosyror än en klassisk trypsin-endast-uppläggning och identifierade fler proteiner vid liknande felnivåer. När det jämfördes direkt med tre ledande databassökningsmotorer fann DiNovo liknande antal aminosyror och proteiner, och de flesta av dess sekvenser överensstämde med dem från sökmotorerna på samma spektra. Författarna menar att denna nivå av täckning och överensstämmelse innebär att de novo-sekvensering, länge behandlad som en reservmetod, nu kan stå sida vid sida med databassökning som ett seriöst och i vissa fall överlägset alternativ.
Större bild: mot fullständig, opartisk läsning av proteiner
För en icke-specialist är slutsatsen att DiNovo gör det mycket lättare att läsa proteindelar exakt utan att vara begränsad till vad som redan finns i referensdatabaser. Genom att fördubbla eller tredubbla mängden välstödd sekvensinformation och erbjuda inbyggda felkontroller öppnar detta tillvägagångssätt dörren för att upptäcka okända proteiner, följa subtila variationer och utforska komplexa blandningar där många komponenter fortfarande är okända. Kort sagt, genom att para ihop spegelenzym med djupinlärning och noggrann statistik hjälper DiNovo att omvandla brusiga spektrala spår till en tydligare och mer tillförlitlig bild av de proteiner som ligger till grund för hälsa och sjukdom.
Citering: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Nyckelord: proteomik, de novo-peptidsekvensering, masspektrometri, djupinlärning, spegelproteaser