Clear Sky Science · sv
Multimodal dissektion av celltyp-specifik TDP-43-patologi i motorcortex
Varför denna forskning betyder något för människor
Amyotrofisk lateralskleros (ALS) och frontotemporal demens (FTD) är förödande hjärnsjukdomar som berövar människor rörelseförmåga, tal och personlighet. De flesta patienter med ALS och många med FTD delar ett mikroskopiskt kännetecken: ansamlingar av ett protein kallat TDP-43 på platser där det inte hör hemma. Denna studie ställer två praktiska frågor med stora konsekvenser för framtida behandlingar: vilka hjärnceller drabbas exakt hårdast av TDP-43-relaterade problem, och vad går fel inne i dessa celler på nivån för DNA-reglering och genaktivitet?
Följa skadan in i hjärnans rörelsencentrum
Forskarna fokuserade på primära motorcortex, det bälte av hjärnvävnad som kontrollerar viljestyrd rörelse. Med hjälp av post mortem-donerade hjärnprover från personer med ALS, ALS-FTD och neurologiskt friska kontroller isolerade de enskilda cellkärnor och läste både vilka gener som var aktiva och hur tätt den lokala DNA-packningen var. Denna “multi-omiska” metod, tillämpad på mer än 180 000 kärnor, gjorde det möjligt att sortera celler i precisa typer: flera klasser av excitatoriska och inhibitoriska neuroner, liksom stödjeceller såsom astrocyter, oligodendrocyter och mikroglia. De kombinerade detta med spatiala genkartor från en annan mänsklig hjärndataset för att placera tillbaka dessa celltyper i cortex välbekanta lagerstruktur.
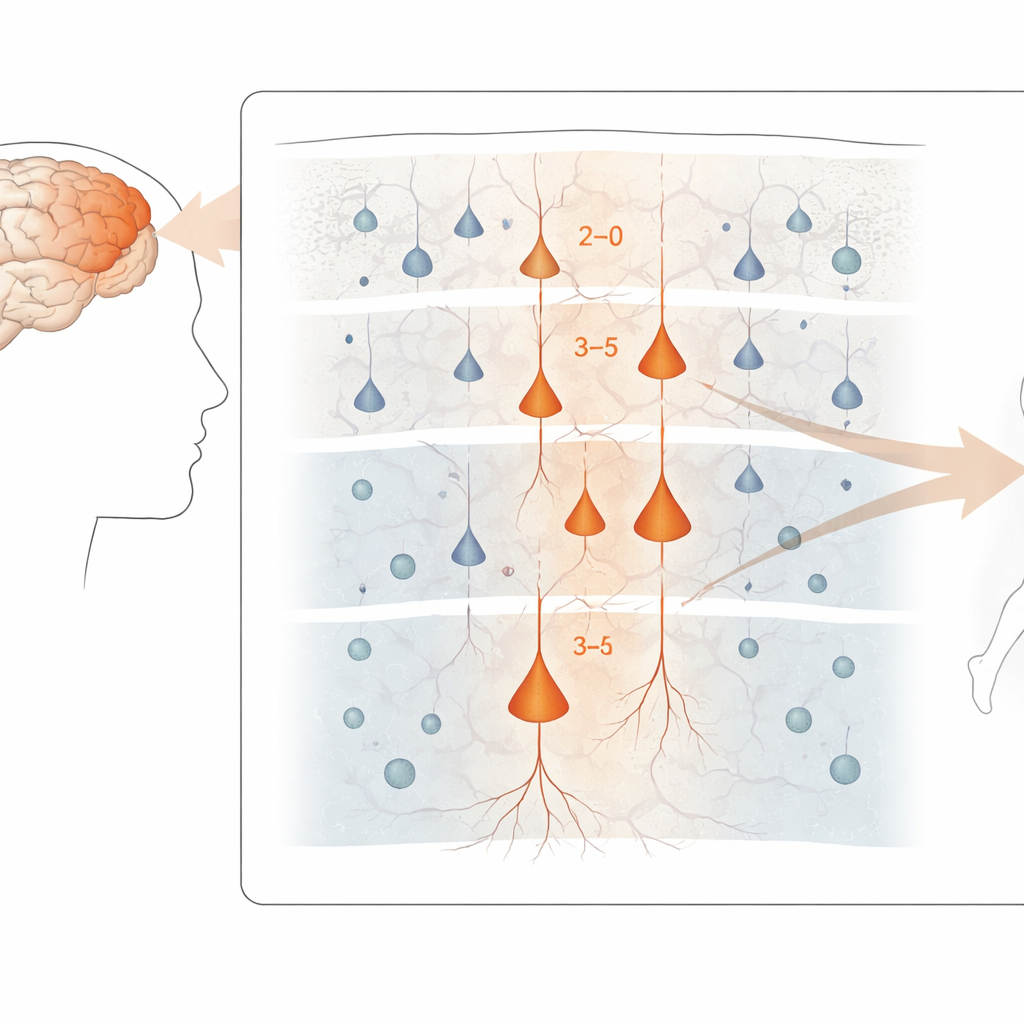
Identifiera de mest sårbara neuronerna
I motorcortex framträdde de starkaste sjukdomsrelaterade genförändringarna i excitatoriska neuroner, cellerna som driver aktivitet framåt i hjärnans kretsar. Särskilt över- och mellanskiktsneuroner som kopplar inom cortex, samt vissa djupa skiktsceller som skickar signaler ut ur cortex — inklusive de stora så kallade Betz-cellerna som kontrollerar ryggmärgens motorneuron — visade de mest uttalade förändringarna. I kontrast påverkades inhibitoriska interneuroner och många gliaceller i mindre grad på genuttrycksnivå, även om vissa av dem visade mer subtila skift. Trots denna molekylära oro var den övergripande sammansättningen av större celltyper i vävnaden förvånansvärt lik mellan patienter och kontroller, vilket tyder på att skadan handlar mer om hur cellerna fungerar än enbart om hur många som gått förlorade.
Hur TDP-43 omformar genaktivitet inifrån
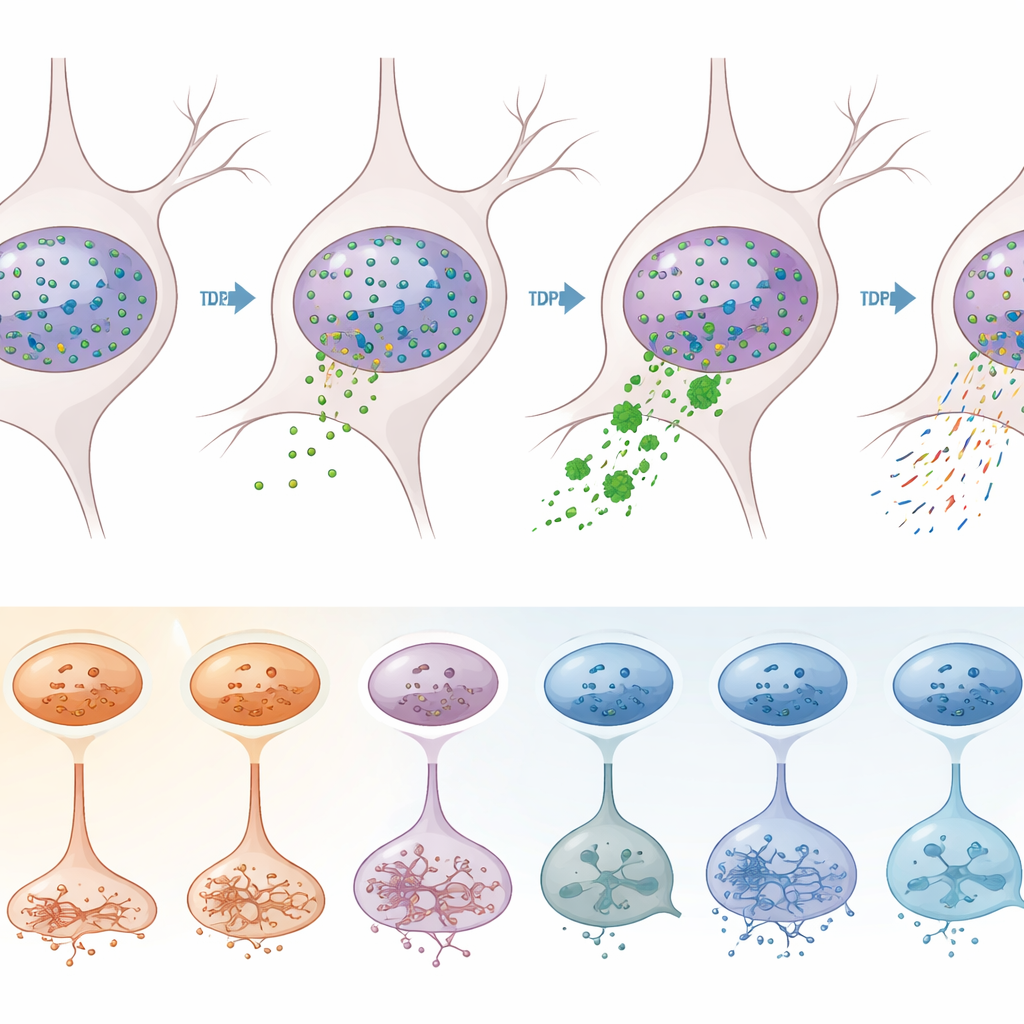
För att separera effekter som drivs direkt av TDP-43 från andra sjukdomsprocesser använde teamet en smart sorteringsstrategi. De märkte kärnorna med antikroppar mot TDP-43 och en neuronal markör, och använde därefter flödescytometri för att skilja neuroner vars kärnor förlorat TDP-43 (en patologisk markör) från dem som behöll det. Sekvensering av mer än 12 000 av dessa kärnor visade att TDP-43-förlusten överväldigande sker i excitatoriska neuroner, särskilt i specifika undertyper i lager 2–3, 3–5, 5 och 6. I dessa sårbara neuroner var hundratals gener felreglerade, inklusive många som redan kopplats till ALS. Klassiska molekylära signaturer för TDP-43-fel — såsom uppkomsten av ’kryptiska’ extra stycken i STMN2- och KALRN-transkript, och skiften i var RNA-molekyler klipps och polyadenyleras i sina ändar — var tydligt berikade i de TDP-43-bristande kärnorna.
Epigenetisk omstrukturering: inte all förändring orsakas av TDP-43
Eftersom de mätte både genaktivitet och kromatinöppenhet i samma kärnor kunde författarna undersöka vilka förändringar som var kopplade till skift i DNA-packning. De hittade tiotusentals platser i genomet där lokal kromatinåtkomlighet följde genuttryck. Många av de gener som förändrades i ALS och ALS-FTD låg i sådana regioner, vilket indikerar att en del av sjukdomssignaturen speglar bredare epigenetisk omstrukturering snarare än direkt följd av TDP-43-förlust. Intressant nog konvergerade dessa kromatinrelaterade förändringar ofta på signalvägar involverade i cellkommunikation och axonvägledning, och de var särskilt starka i vissa excitatoriska neuroner och oligodendrocyter. När teamet jämförde genförändringar kopplade till TDP-43-patologi med de kopplade till kromatinförskjutningar såg de att dessa delvis överlappade men till stor del utgjorde skilda lager av störning.
Vad detta betyder för framtida terapier
För en lekmannaläsare är huvudbudskapet att ALS och ALS-FTD inte skadar motorcortex jämnt. Istället drabbar de särskilda typer av excitatoriska neuroner och, i mindre grad, vissa stödjeceller, och ändrar deras genprogram på sätt som beror både på TDP-43:s misskötsel och på bredare förändringar i hur DNA packas och avläses. Dessa fynd tyder på att effektiva behandlingar kan behöva vara både celltypsspecifika och vägspesifika — till exempel återställa TDP-43-funktion eller korrigera dess splitsningsfel i de mest sårbara neuronerna, samtidigt som man separat riktar in sig på epigenetiska och signaleringsförändringar som delas av flera celltyper. Genom att kartlägga detta komplexa landskap i hög detalj ger studien en ritning för att utforma mer precisa interventioner med målet att bromsa eller förhindra förlusten av rörelsekontroll vid ALS och ALS-FTD.
Citering: Ruf, W.P., Kühlwein, J.K., Meier, L. et al. Multi-modal dissection of cell-type specific TDP-43 pathology in the motor cortex. Nat Commun 17, 2406 (2026). https://doi.org/10.1038/s41467-026-69944-6
Nyckelord: ALS, frontotemporal demens, TDP-43, motorcortexneuroner, single-nucleus multiomics