Clear Sky Science · sv
Effektiv provtagning av storskaliga övergångsvägar och intermediära konformationer i sub-mesoskopiska proteinkomplex
Att se proteiner i rörelse
Många av de molekyler som håller oss vid liv beter sig mindre som styva legobitar och mer som små maskiner som ständigt ändrar form. Dessa rörelser driver processer som energiproduktion, DNA-reparation och hur virus tar sig in i celler. Experiment som kryo-elektronmikroskopi kan nu frysa några av dessa former, men inte de flyktiga stegen däremellan. Den här artikeln presenterar eBDIMS2, en ny datormetod som kan ”fylla i de saknade bilderna” av proteinrörelser även för enorma molekylära maskiner som tidigare var för stora och komplexa för att simulera på en vanlig dator.
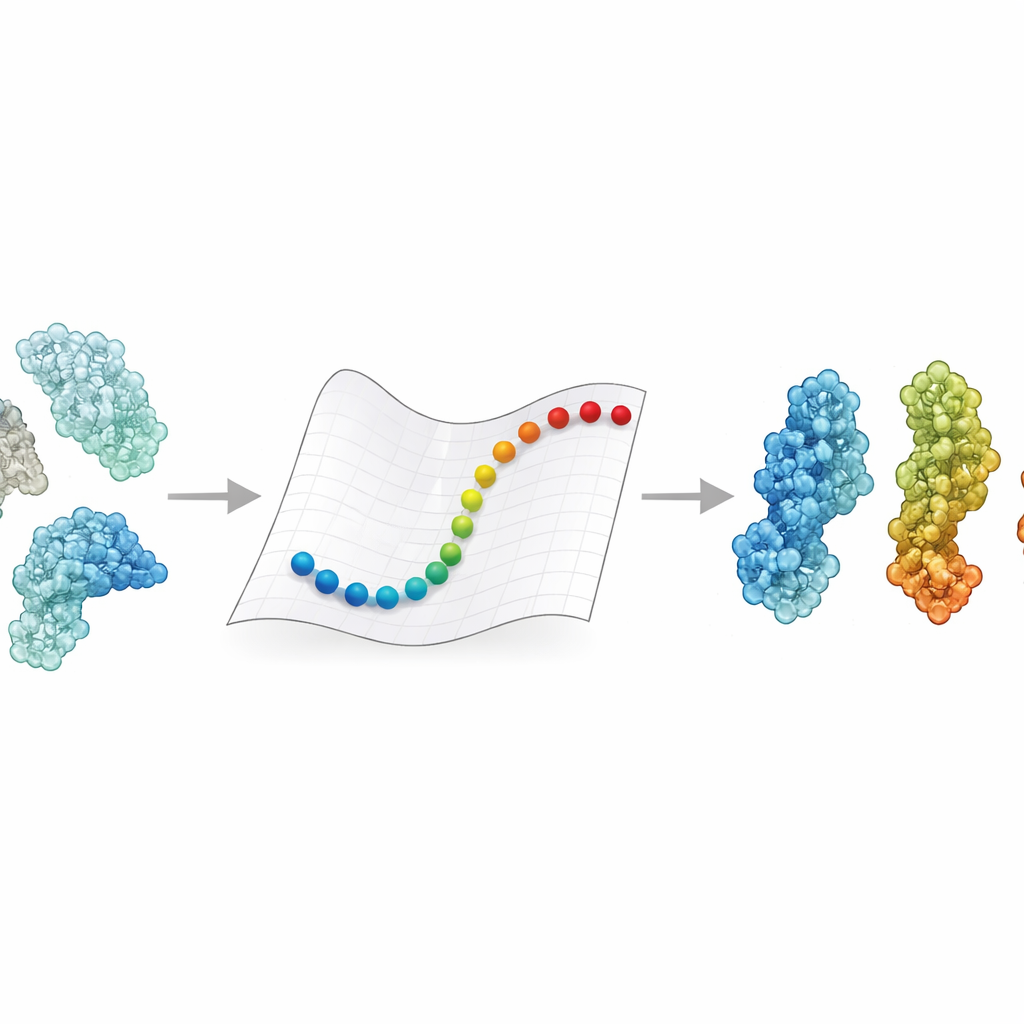
Varför proteins formförändringar spelar roll
Proteiner förblir sällan låsta i en enda ställning. De öppnar och stänger sig, vrider och böjer sig som svar på signaler som spänningsförändringar, pH eller bindning av en partnermolekyl. Dessa skift kan avgöra om ett enzym är aktivt eller inte, eller om en receptor fångar ett virus eller låter det glida förbi. Experiment ger oss detaljerade ögonblicksbilder av ett fåtal nyckelformer, och molekylär dynamik kan i princip koppla ihop dem genom att följa varje atom över tid. Men att spåra sådan rörelse för de enorma komplex som nu ses med kryo-elektronmikroskopi — ofta med massor på hundratusentals till miljontals dalton — kräver vanligtvis superdatorer och veckor av beräkningar. Som en följd vet vi fortfarande inte för många medicinskt viktiga jättar hur ett tillstånd omvandlas till ett annat.
En snabbare väg genom proteinlandskap
eBDIMS2 tar en genväg genom att förenkla hur proteiner representeras och hur deras rörelse beräknas. Istället för att följa varje atom behandlar metoden varje aminosyra som en enda punkt som kopplas samman med fjädrar i ett elastiskt nätverk. Dessa fjädrar fångar hur olika delar av proteinet tenderar att röra sig tillsammans. Metoden använder sedan Brownsk dynamik — matematiska regler som efterliknar studsandet i en vätska — för att skjuta strukturen från ett experimentellt känt tillstånd mot ett annat. Viktigt är att eBDIMS2 bara uppmärksammar de interaktioner som verkligen betyder något, och använder avståndsgränser samt parallellberäkning för att minska kostnaden. Det förbättrar programmets skalning från ungefär kvadratisk till nästan linjär med proteinstorlek. I praktiken innebär det att övergångar för enorma uppsättningar som närmar sig två miljoner dalton kan utforskas på timmar på en stationär dator, istället för att vara i princip otillgängliga.
Jämförelse av vägarna med riktiga proteiner
För att avgöra om dessa snabba vägar är biologiskt meningsfulla samlade författarna en ensemble av 47 stora proteiner och 15 ytterligare komplex, totalt hundratals strukturer främst lösta med kryo-elektronmikroskopi. De använde huvudkomponentsanalys, ett statistiskt verktyg som fångar de dominerande sätten ett protein kan röra sig på, för att organisera dessa strukturer i konformationslandskap som öppet, stängt, aktivt eller inaktivt. eBDIMS2 fick sedan i uppdrag att koppla ihop par av ändtillstånd över detta landskap. De resulterande vägarna projicerades tillbaka på samma lågdimensionella kartor för att avslöja om de följer släta rutter som passerar nära experimentellt observerade intermediärer. I mer än 30 % av systemen gick de simulerade rutterna nära — inom några ångström — intermediära strukturer som inte hade lämnats in som input. För krävande fall som DNA-reparationsenzymet DNA-PKcs eller koronavirusets spike-protein överensstämde de grovkorniga vägarna också väl med mycket dyrare atomnivåsimuleringar, inklusive riktad molekylär dynamik och avancerade metoder för förbättrad sampling.
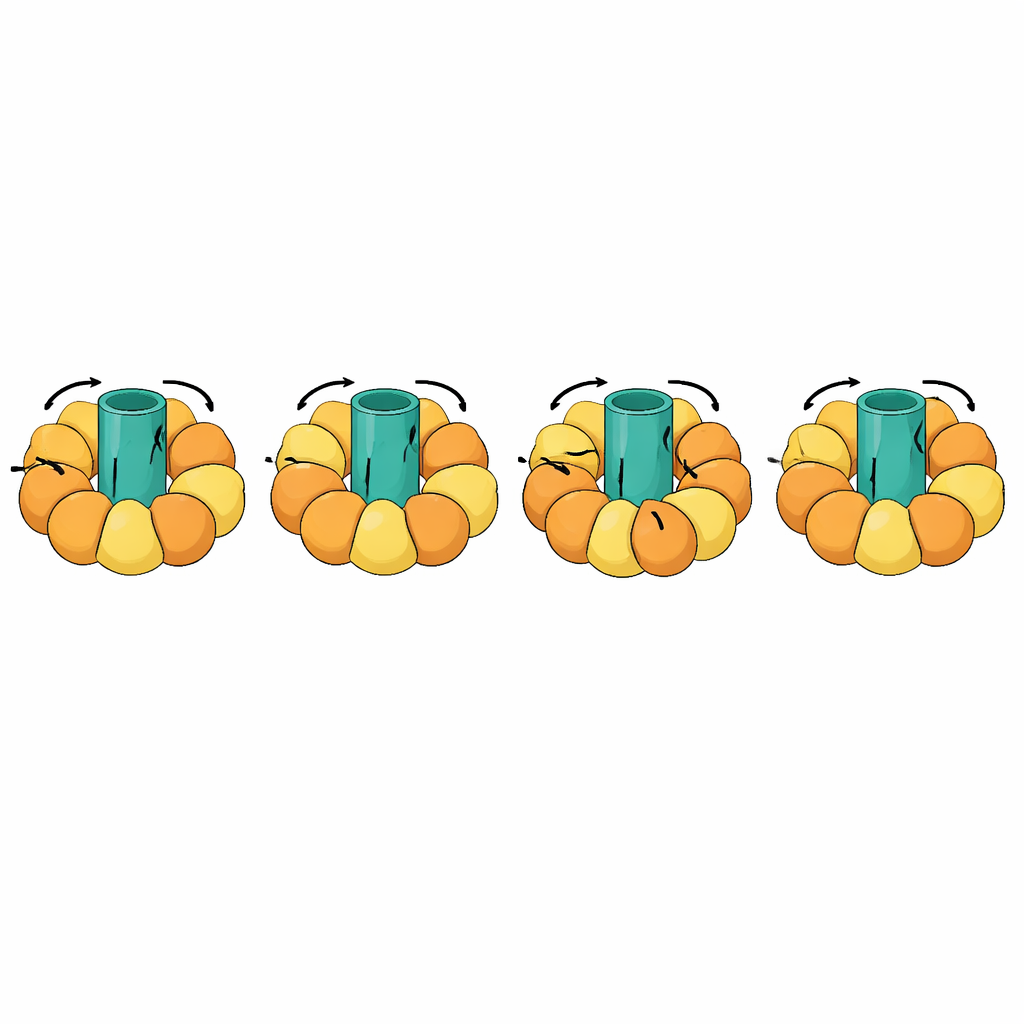
Att följa jättelika molekylära maskiner
Ett av de mest slående testerna omfattade roterande maskiner som ATP-syntaser, som producerar cellens energivaluta genom att koppla en snurrande rotor i membranet till öppnings- och stängningsrörelser i omgivande subenheter. Dessa övergångar är extremt komplexa: delar av molekylen måste förbli styva och rotera som en enhet, medan andra flexar i en koreograferad cykel. eBDIMS2 inför särskild hantering för sådana kvasi-styva delar och för ofullständiga experimentella modeller med saknade segment, vilket är vanligt i kryo-elektronmikroskopi. Med dessa funktioner kan den simulera fullständiga rotationscykler för ATP-syntas och andra massiva komplex som molekylära chaperoner, receptorer och virusuppbyggnader. Genomgående undviker de genererade intermediära strukturerna allvarliga deformationer som vissa konkurrerande metoder framkallar och kan sedan rengöras till atomistiska modeller som lämpar sig för läkemedelsdesignberäkningar eller längre, mer detaljerade simuleringar.
Vad detta betyder för biologi och medicin
Studien visar att eBDIMS2 på ett tillförlitligt sätt kan skissa huvudvägarna mellan kända proteinformer för system som tidigare varit utom räckhåll för traditionella simuleringar. Den ersätter inte detaljerade atomnivåfilmer eller ger exakta energier och tidsskala, men den erbjuder ett snabbt, fysikaliskt grundat sätt att kartlägga hur stora molekylära maskiner kan röra sig med endast ett par experimentella strukturer som input. När strukturdatasamlingar fylls med flera tillstånd av stora proteinkomplex kopplade till cancer, infektion och andra sjukdomar, ger detta tillvägagångssätt forskare ett lättillgängligt verktyg för att koppla ihop punkterna, föreslå sannolika intermediära tillstånd och vägleda var man bör söka vidare med högre upplösning eller riktad läkemedelsdesign.
Citering: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Nyckelord: proteindynamik, molekylära simuleringar, cryo-EM, konformationsvägar, grovkornig modellering