Clear Sky Science · sv
Aktivering av IRF3 i kardiomyocyter försämrar mitokondriell oxidativ funktion genom hämning av PGC-1α och driver hjärtsvikt
Varför stressade hjärtan och utmattade celler spelar roll
Hjärtsvikt beskrivs ofta som att hjärtat ”slits ut”, men under ytan är det också en berättelse om kronisk inflammation och utarmade kraftverk inne i hjärtmuskelcellerna. Denna studie ställer en till synes enkel fråga med stora konsekvenser: finns det en enda molekylär strömbrytare i hjärtceller som knyter ihop skadlig inflammation och fallerande energiproduktion — och om så är fallet, kan man förändra sjukdomsförloppet genom att påverka den? Genom att följa den tråden hittar författarna en nyckelspelare och visar att en måttlig uppstärkning av hjärtats eget energiprogram delvis kan rädda sviktande hjärtan hos möss.
En molekylär strömbrytare i sjuka mänskliga hjärtan
Forskarna fokuserade på ett protein kallat IRF3, mest känt för att hjälpa celler reagera på virusinfektioner. De undersökte vävnad från personer med ischemisk kardiomyopati, en vanlig form av hjärtsvikt orsakad av minskat blodflöde efter hjärtinfarkter. I dessa sviktande hjärtan var IRF3 inte bara närvarande — det var kemiskt aktiverat på specifika platser, ett tecken på att det aktivt drev genprogram. Samtidigt var de maskiner som låter mitokondrier omvandla bränsle till energi via oxidativ fosforylering märkbart försvagade. Ett liknande mönster sågs i musmodeller av hjärtinfarkt: när en kranskärl förbandes aktiverades IRF3 kraftigt i hjärtmuskelceller, och gener styrda av IRF3 tändes. Till och med fragment av mitokondriellt DNA — frigjort från skadade mitokondrier och agerande som interna ”fara”-signaler — räckte för att slå på IRF3 i isolerade hjärtceller.
Att stänga av IRF3 skyddar hjärtat
För att testa om IRF3-aktivitet i hjärtmuskelceller faktiskt förvärrar sjukdomen konstruerade teamet möss där IRF3 kunde tas bort endast från kardiomyocyter, medan andra immunceller och stödjeceller lämnades orörda. Efter att ha framkallat en hjärtinfarkt hade dessa möss bättre pumpförmåga och mindre ärrbildning än normala möss, trots samma initiala skada. I hjärtceller odlade i laboratorieplattor dämpade tystande av IRF3 inflammatoriska gener utan att störa andra närliggande proteiner. Tillsammans talar dessa resultat för att IRF3 inne i hjärtcellen själv inte bara är en åskådare: det förstärker inflammation och strukturell skada efter ischemi och hjälper till att driva övergången till hjärtsvikt.
När IRF3 sitter fast i "på"-läge kollapsar bränslesystemet
Författarna vände sedan experimentet: de skapade möss där IRF3 i kardiomyocyter kunde tvingas in i ett permanent aktivt tillstånd med en smart genetisk ”fosfomimetisk” lösning. Även utan en yttre trigger utvecklade dessa möss snabbt svår hjärtdysfunktion, höga nivåer av inflammatoriska budbärare i blodet och tecken på cellskada. En djupgående analys av deras hjärtvävnad visade att när IRF3 är kroniskt aktivt så undertrycker det en huvudregulator för energi kallad PGC-1α. Denna molekyl främjar normalt friska mitokondrier, effektiv förbränning av fetter och balanserad cellulär energi. När PGC-1α trycks ner föll flera mitokondriella proteiner, elektrontransportkedjan sviktade och hjärtats bränsleval förändrades: karnitin och relaterade föreningar för fettförbränning minskade, ketonanvändning var nedsatt och glukosmetabolismen blev rubbad. Även kvoten NAD⁺/NADH — en viktig indikator på cellens redoxbalans — försköts åt fel håll.
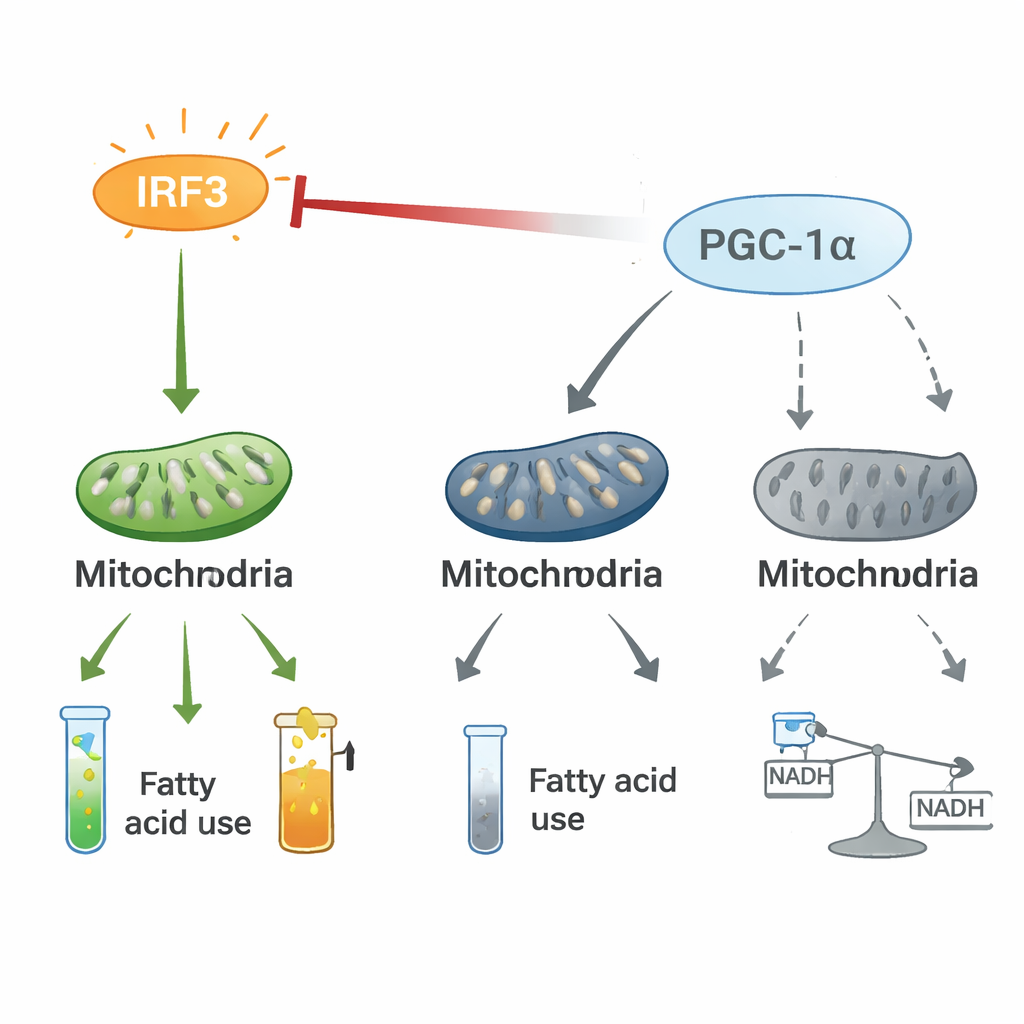
En dragkamp mellan inflammation och energikontroll
Mekanistiska experiment visade att IRF3 och PGC-1α bildar en tvåvägsreglerande axel. I hjärtceller associerar aktiverat IRF3 fysiskt med PGC-1α och dämpar dess förmåga att slå på gener för fettförbränning. Nedtystning av IRF3 höjer PGC-1α-nivåer och aktivitet, medan att öka PGC-1α dämpar IRF3-driven inflammatorisk genaktivitet och återställer mitokondriella markörer, även under stressförhållanden som lågt syre eller bakteriella toxiner. Stabil isotopspårning visade att IRF3-aktivering omdirigerar kol från normal energiproduktion via citronsyracykeln in i en alternativ väg, pentosfosfatvägen, och stör det jämna flödet av metaboliter. Denna dragkamp mellan en proinflammatorisk strömbrytare (IRF3) och en energikoordinator (PGC-1α) verkar omforma hjärtats metabolism på sätt som gynnar inflammation och energiförluster.
Måttlig återuppladdning av hjärtats batterier
Slutligen frågade teamet om en lätt höjning av PGC-1α skulle kunna motverka IRF3:s skada. De använde en hjärt-riktad genterapivektor för att måttligt — men inte överdrivet — öka PGC-1α i samma möss med hyperaktivt IRF3. Denna blygsamma ökning förbättrade pumpfunktionen, ökade mitokondriella proteiner, förstärkte gener för fettförbränning och NAD-metabolism samt minskade inflammatorisk och fibrotisk genaktivitet. I cellexperiment återställde samtidig uttryck av PGC-1α med aktivt IRF3 en hälsosammare NAD⁺/NADH-balans och skiftade bränsleanvändningen tillbaka mot fetter. För en lekmannaläsare innebär detta att en försiktig återuppladdning av hjärtats ”batterihanteringssystem” delvis kan kompensera de skadliga effekterna av en kronisk inflammatorisk strömbrytare som sitter i "på"-läge.
Vad detta betyder för framtida hjärtsviktsvård
Detta arbete placerar IRF3 som en central länk mellan inflammation och energisvikt inne i hjärtmuskelceller. Istället för att behandla inflammation och metabolism som separata problem vid hjärtsvikt föreslår studien att de är sammanflätade genom en IRF3–PGC-1α-axel. Även om dessa fynd är gjorda i möss och celler öppnar de möjligheten att framtida behandlingar antingen kan dämpa IRF3-aktivitet eller stärka PGC-1α och mitokondriell funktion för att bromsa eller förebygga hjärtsvikt efter en hjärtinfarkt. Enkelt uttryckt kan en lugnning av ett överaktivt cellulärt alarmsystem tillsammans med stöd för hjärtats energifabriker visa sig vara en kraftfull, kombinerad strategi för att hålla försvagade hjärtan igång längre.
Citering: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Nyckelord: hjärtsvikt, inflammation, mitokondrier, kardiomyocyter, PGC-1α