Clear Sky Science · sv
Att utöka räckvidden för grafneuronnät med globala kodningar
Varför långdistanskopplingar i molekyler spelar roll
Från nya läkemedel till bättre batterier bygger många av dagens genombrott på datormodeller som kan förutsäga hur tusentals atomer drar i och påverkar varandra. En populär klass av AI-modeller, kallad grafneuronnät, har blivit ett arbetsredskap för denna uppgift. Men dessa modeller har en blind fläck: de fokuserar mest på nära grannar, trots att avlägsna atomer kan påverka varandra starkt genom elektriska och kvantmekaniska krafter. Denna artikel presenterar RANGE, ett sätt att ge dessa neuronnät en slags global överblick så att de kan ”känna” och förutsäga långdistanseffekter utan att bli smärtsamt långsamma eller minneskrävande.
Hur dagens AI bara ser grannskapet
Grafneuronnät behandlar en molekyl eller ett material som ett nät av noder (atomer) förbundna av kanter (deras relationer). Vid varje steg uppdaterar varje nod sitt tillstånd genom att kommunicera endast med sina närliggande grannar inom en fast radie. Genom att upprepa detta sprids information långsamt, men denna strategi har två stora nackdelar. För det första kan meddelanden suddas ut när de passerar genom många mellanhänder, ett problem som kallas oversmoothing. För det andra kan smala informationsvägar i grafen begränsa hur mycket information som kommer igenom, vilket leder till oversquashing. Båda problemen blir allvarliga när man försöker fånga krafter som verkar över många ångström, såsom elektrostatik och dispersion i stora molekyler eller kristaller. Att helt enkelt förlänga interaktionsavståndet eller stapla fler lager gör modellerna dyrare och löser inte fullt ut dessa flaskhalsar.
Lägga till virtuella nav för global kommunikation
RANGE (Relaying Attention Nodes for Global Encoding) omtecknar denna bild genom att lägga till en liten mängd virtuella ”master-noder” som inte motsvarar några verkliga atomer. Istället fungerar de som globala nav. Efter ett vanligt meddelande-passeringssteg mellan närliggande atomer samlas information från alla atomer in i dessa nav med hjälp av en attention-mekanism: varje master-nod lär sig vilka delar av systemet den ska fokusera på. Denna aggregering skapar grovkorniga sammanfattningar av molekylens tillstånd. I ett andra sändningssteg skickas dessa sammanfattningar tillbaka till varje atom, återigen med attention så att varje atom kan avgöra hur mycket den ska lyssna på varje master-nod samtidigt som den behåller sitt lokala minne via självreferenser. Eftersom varje atom är direkt kopplad till varje master-nod kan långdistanskommunikation ske i ett enda steg, vilket förvandlar grafen till ett small-world-nätverk där avlägsna regioner snabbt och effektivt kan påverka varandra. 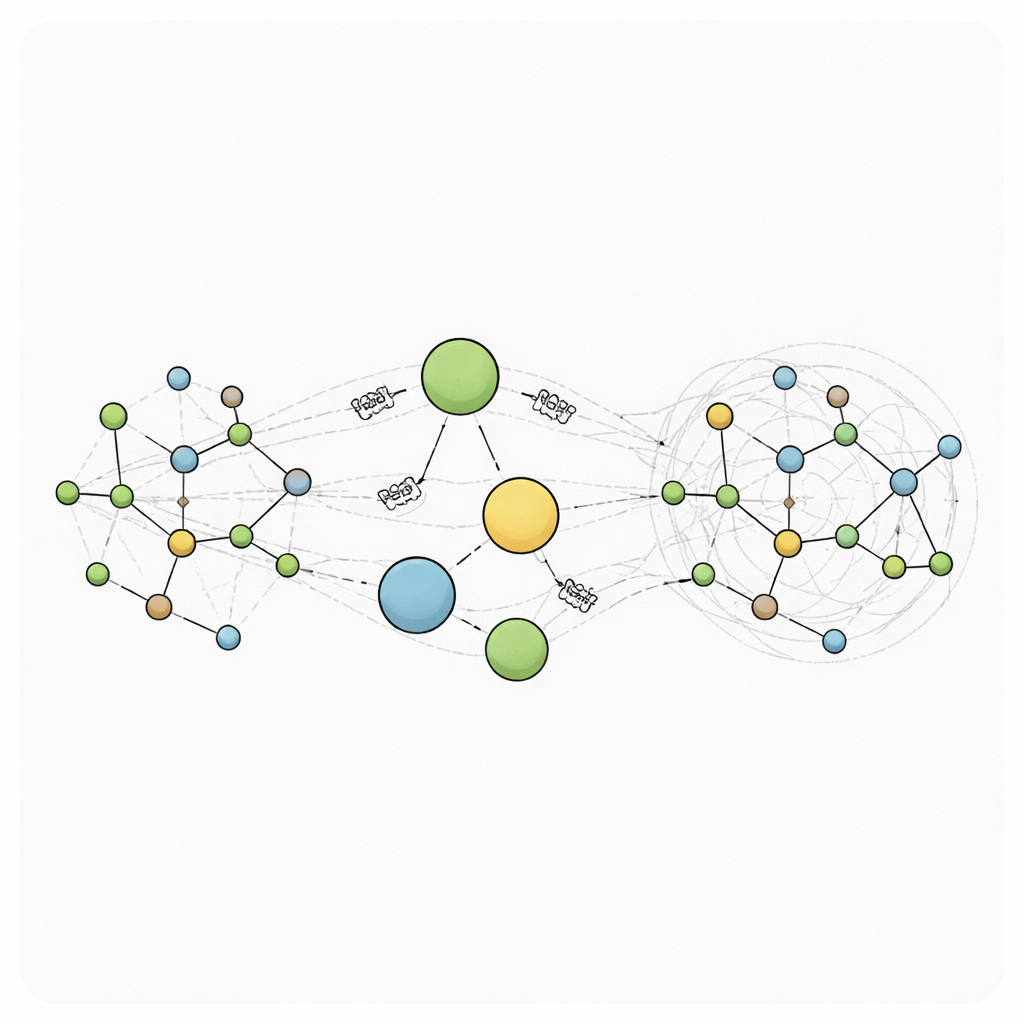
Upptäcka långdistanskrafter som andra missar
Forskarna testade RANGE genom att koppla det till flera toppmoderna molekylära kraftfältsmodeller och jämföra dem med deras ursprungliga, rent lokala versioner. De använde utmanande system där långdistans-effekter är kända för att vara avgörande: en salts kristall med en extra natriumjon som fungerar som dopant, en gulddimer som närmar sig en dopad oxidyta, och par av organiska molekyler som interagerar på varierande avstånd. Standardmodeller missade till stor del hur avlägsna laddningsomfördelningar eller dolda dopanter förändrade energilandskapet; deras förutsägelser förändrades knappt när den långväga omgivningen ändrades. Däremot fångade RANGE-augmenterade modeller korrekt de olika energikurvorna och kunde extrapolera till större separationer än de sett under träning, med fel upp till fyra gånger mindre för svåra laddade dimerer.
Noggrannhet utan att krossa datorn
Avgörande är att RANGE ger denna förbättrade överblick utan den branta beräkningskostnaden hos andra globala metoder. Tekniker som lånar från fysiken, som Ewald-summation eller Fourier-baserade korrigeringar, kräver operationer som växer ungefär med kvadraten på antalet atomer eller är beroende av stora galler, vilket gör dem tunga för stora system och upprepade simuleringar. RANGE, till sin natur, skalar linjärt med systemstorleken: varje master-nod kopplas till alla atomer, men antalet master-noder växer måttligt och styrs av en regulariseringsmetod som förhindrar att de blir överflödiga. Benchmarktester på större datamängder visar att RANGE konsekvent minskar felen i förutsagda krafter, även när de underliggande modellerna använder korta interaktionsgränser, och det görs med endast en måttlig ökning i körtid och minnesanvändning. Teamet körde också molekyldynamiksimuleringar över tiotals nanosekunder på komplexa molekyler och fann att RANGE-baserade kraftfält förblev stabila och utforskade realistiska former och tillstånd. 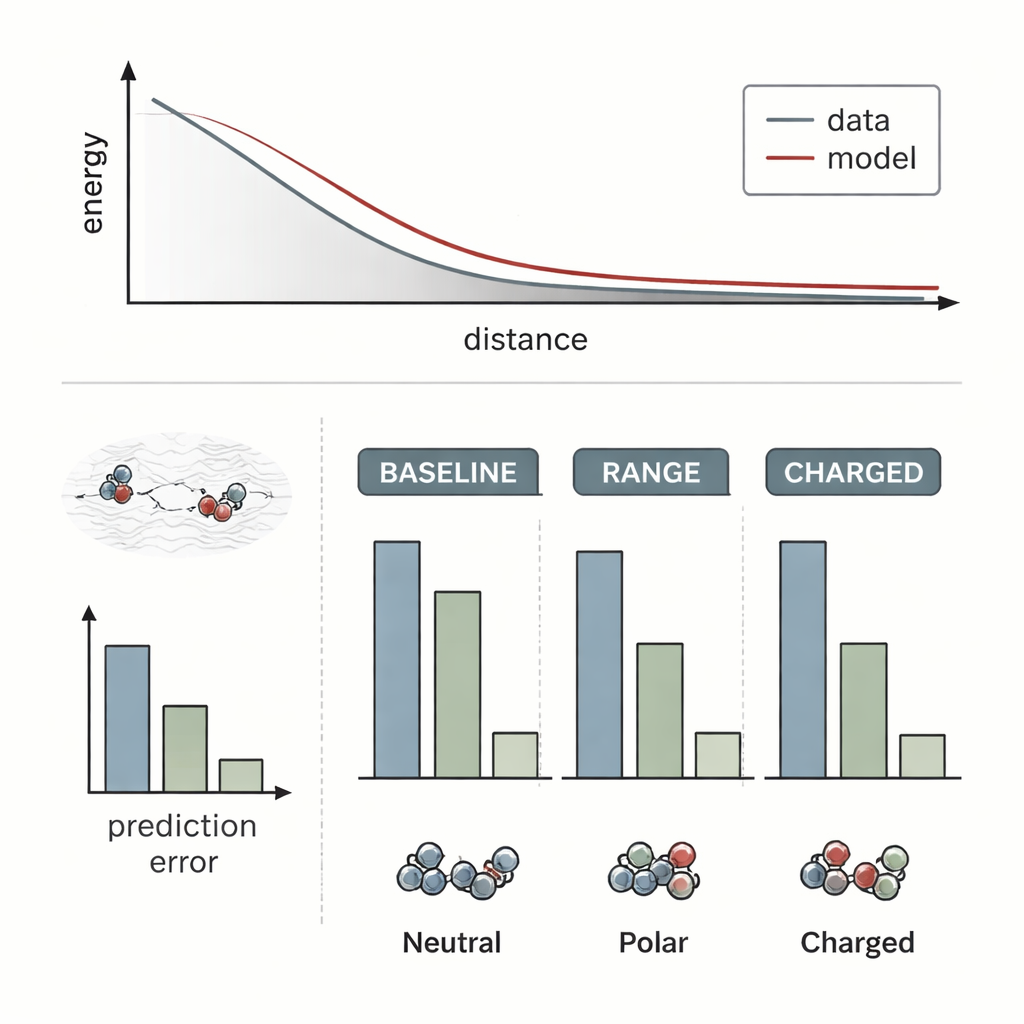
En tydligare helhetsbild av molekylära världar
För icke-specialister är huvudbudskapet att RANGE ger befintliga grafbaserade AI-modeller ett nytt sätt att tänka globalt samtidigt som de fortfarande verkar lokalt. Genom att införa intelligenta virtuella nav och attention-driven informationsflöde övervinner det de vanliga flaskhalsarna som hindrar neuronnät från att fånga långdistans-, många-kropps-effekter i molekyler och material. Det innebär mer pålitliga förutsägelser för system där avlägsna regioner subtilt påverkar varandra — från flexibla läkemedelsmolekyler till utsträckta nanostrukturer — utan en förödande beräkningskostnad. När dessa metoder tillämpas på allt större och mer komplexa miljöer lovar de AI-verktyg som bättre kan spegla den verkliga, långdistanspräglade väven av fysiska interaktioner.
Citering: Caruso, A., Venturin, J., Giambagli, L. et al. Extending the range of graph neural networks with global encodings. Nat Commun 17, 1855 (2026). https://doi.org/10.1038/s41467-026-69715-3
Nyckelord: grafneuronnät, långdistansinteraktioner, molekylära simuleringar, maskininlärda kraftfält, attention-mekanismer