Clear Sky Science · sv
Patofysiologisk betydelse av försämrad KAT7‑beroende histon H3K14‑acetylering vid zinkbrist
Varför små näringsämnen spelar roll för vår hälsa
Zink är en spårmetall som våra kroppar bara behöver i små mängder, men som tyst stödjer hundratals proteiner som håller cellerna igång. När zink saknas—på grund av kost, sjukdom eller åldrande—har det kopplats till problem från dålig tillväxt till nedsatt immunförsvar och fettlever. Denna studie ställer en djupare fråga: hur känner cellerna faktiskt av att zink blir knapp, och hur kan denna brist översättas till bestående förändringar i genaktivitet och organhälsa?
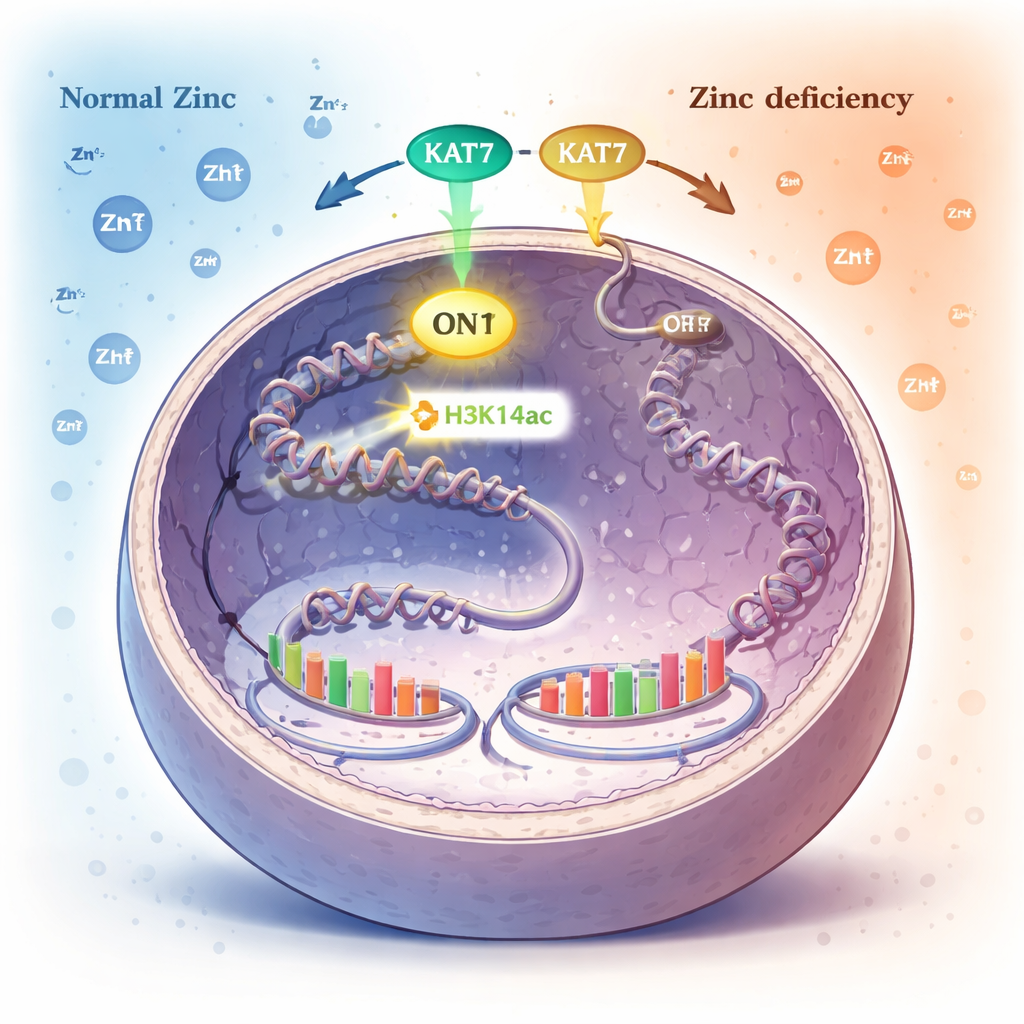
En kemisk markör på DNA‑paketeringen som en intern zinklarm
Inne i kärnan är DNA lindat runt proteinspolar kallade histoner. Celler kontrollerar vilka gener som är aktiva genom att lägga till eller ta bort små kemiska markörer på dessa histoner. En sådan markör, acetylering på en specifik plats på histon H3 (H3K14ac), läggs på av ett enzym som heter KAT7. Författarna upptäckte att när zink blir knapp sjunker nivåerna av denna H3K14ac‑markör dramatiskt, medan många andra vanliga histonmarkörer förblir oförändrade. Detta pekade ut H3K14ac, och enzymet KAT7 som skapar den, som en nyckelsensor för zinkstatus.
Hur zink håller ett viktigt enzym påslaget
Genom att systematiskt inaktivera olika enzymer visade forskarna att KAT7 är huvudkällan till H3K14ac i mänskliga celler. KAT7 innehåller en liten zinkbindande struktur i sitt aktiva centrum. När celler pressades in i zinkbrist minskade KAT7:s förmåga att sätta H3K14ac‑märket, även om proteinet självt stannade i kärnan och förblev associerat med sina medhjälparpartner. Detaljerade tester med renade KAT7‑fragment visade att korrekt bunden zink i detta område är avgörande för dess aktivitet; att störa zinkbindningen slog av enzymet, och att noggrant tillsätta zink återställde funktionen. I praktiken beter sig KAT7 som en zinkberoende brytare som kontrollerar en specifik histonmarkör.
Att omvandla zinkförlust till genförändringar som återställer zinknivåer
Vad gör förlusten av denna histonmarkör i praktiken? Med hjälp av genomomfattande kartläggning visade teamet att H3K14ac är särskilt rikt vid enhancer‑regioner—regulatoriska DNA‑sträckor som finjusterar närliggande gener. Under zinkbrist togs H3K14ac bort från många enhancers, och ju större förlusten var desto starkare förändring i intilliggande genaktivitet observerades. En framträdande gen var ZIP10, som kodar för ett protein i cellmembranet som importerar zink. När H3K14ac föll vid ZIP10:s enhancer ökade ZIP10‑nivåerna på membranet, vilket tillät mer zink att strömma in i cellen. Att blockera KAT7 eller förhindra borttagandet av H3K14ac störde denna respons och minskade zinkupptaget, även efter att zink tillsattes igen. Detta visar att celler omvandlar zinkbrist till en epigenetisk signal som förstärker zinkimportmaskineriet för att återställa balansen.
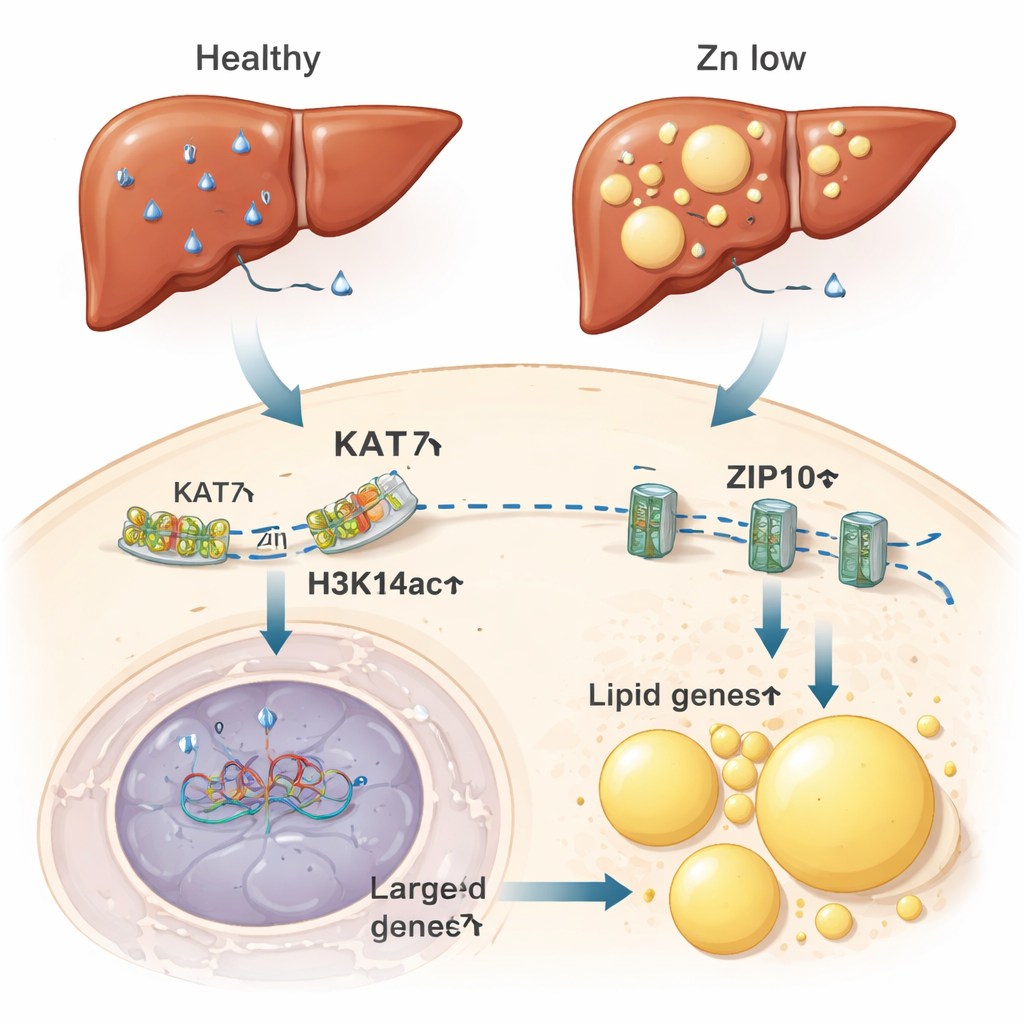
Från zinkutsvultna celler till feta leverar
Författarna frågade sedan om denna zinkkänsliga brytare har konsekvenser i hela djur. Hos möss som matades med en zinkfattig diet visade levern—en central knutpunkt för zink‑ och fettmetabolism—minskade zinknivåer, lägre H3K14ac och försvagad KAT7‑aktivitet. Dessa förändringar sammanföll med högre uttryck av gener som driver fettinlagring och bildandet av lipidkroppar, de mikroskopiska fettpaketen inne i cellerna. Levrarna hos zinkbristmöss ackumulerade fett i en grad jämförbar med möss på en högfettkost. Anmärkningsvärt nog räckte det att sänka KAT7‑aktiviteten med ett läkemedel, även utan att ändra dietärt zink, för att främja fettuppbyggnad i leverceller. Omvänt minskade tillsatt extra zink fettoxidationen orsakad av en högfettdiet.
Vad detta betyder för risken för sjukdom hos människor
För att sätta sina fynd i kliniskt sammanhang granskade forskarna studier på människor som mätt zinknivåer i levervävnad. I flera rapporter hade personer med fettlever och relaterade störningar signifikant mindre zink i levern än friska kontroller. Tillsammans med musexperimenten tyder detta på att kronisk zinkbrist kan främja fettlever genom att stänga av KAT7, radera H3K14ac‑märket och bestående öka uttrycket av gener som gynnar fettinlagring. Enkelt uttryckt visar arbetet en intern ”zink‑till‑epigenetik” krets: när zink faller mister ett zinkberoende enzym kraft, vilket ändrar DNA‑paketeringen på sätt som först hjälper celler att ta in mer zink, men med tiden också kan driva levern mot ohälsosam fettansamling.
Citering: Fujisawa, T., Takenaka, S., Maekawa, L. et al. Pathophysiological significance of impaired KAT7-dependent histone H3K14 acetylation during zinc deficiency. Nat Commun 17, 1710 (2026). https://doi.org/10.1038/s41467-026-69476-z
Nyckelord: zinkbrist, epigenetik, leverfett, histonacetylering, zinktransportörer