Clear Sky Science · sv
Kan ferric-oxyl-exciterade tillstånd förklara förlängda järn-syrebindningar i heme-peroxidasens katalytiska intermediärer?
Varför järn–syrebindningar i enzymer spelar roll
Inuti våra celler använder särskilda proteiner, enzymer, syre för att på ett säkert sätt utföra kraftfulla kemiska reaktioner. Bland dem förlitar sig heme-peroxidaser på ett järn–syre-par i sitt centrum för att bryta ned väteperoxid, en reaktiv och potentiellt skadlig molekyl. I årtionden har forskare varit oense om den exakta naturen hos denna järn–syrebindning: är den mer lik en stark dubbelbindning eller en lösare enkelbindning — och vad innebär det för hur dessa enzymer fungerar? Den här studien tar sig an den gåtan med ultraska röntgenmetoder och avancerade beräkningar och visar att svaret ligger i flyktiga exciterade tillstånd i själva järn–syre-enheten.
Följa ett enzym i realtid
Forskarna fokuserade på en bakteriell färgavblekande peroxidas, ett heme-enzym som normalt cyklar genom två viktiga högenergiformer, kända som Compound I och Compound II. Dessa former innehåller båda ett järn som binder syre och är centrala för hur enzymet hanterar väteperoxid och oxiderar andra molekyler. Tidigare experiment på liknande enzymer gav förbryllande långa järn–syrebindningslängder, vilket några forskare tolkade som att den antagna mycket reaktiva järn–syre-enheten antingen hade påverkats av röntgenstrålning eller hade plockat upp en extra proton, vilket ändrade dess karaktär. För att undvika sådana artefakter använde teamet tidsupplöst seriefemtosekund-röntgenkristallografi vid en röntgenfria-elektronlaser och fångade diffraktions- och röntgenemissionssignaler från tusentals små proteinkristaller vid rumstemperatur, alla inom tiotals femtosekunder — snabbare än skada hinner uppstå.
Se kemin utvecklas inne i kristallerna
I deras uppställning blandades mikrokristaller av en något modifierad version av enzymet med väteperoxid direkt på ett rörligt band och undersöktes sedan efter fördröjningar från en halv sekund till tiotals minuter. Tidiga tidpunkter gynnade bildandet av Compound I, medan senare tidpunkter dominerades av Compound II. Strukturella data visade att i båda intermediärerna sitter järnatomen intill en enskild syreatom i heme-fickan, och att skyddande loopregioner i proteinet förskjuts för att skärma av detta starkt oxiderande centrum. Viktigt nog visade precisa mätningar att järn–syrebindningens längd låg runt 1,83 ångström vid alla tidpunkter — längre än väntat för en klassisk dubbelbundet ferryl (Fe(IV)=O)-art och närmare en enkelbindning — ändå indikerade spektrala signaturer från röntgenemission och optiskt absorption tydligt höga oxidationsstater i linje med Compounds I och II.
Utesluter enkla förklaringar
Eftersom experimenten utfördes med ultrakorta pulser vid rumstemperatur kunde de vanliga misstänkta orsakerna till förvrängda bindningslängder — röntgeninducerad reduktion och kryogena artefakter — i stor utsträckning avfärdas. Teamet testade också om syret bundet till järnet hade protonerats och förvandlat dubbelbindningen till en hydroxidliknande enkelbindning. Dock talar kända syra–bas-egenskaper hos liknande heme-center, tillsammans med tidigare kemiska studier, starkt emot sådan protonering i denna typ av enzym. De spektroskopiska data visade dessutom att järnet förblev i en hög oksidations- och lågspinntilstånd efter reaktion med väteperoxid, precis som väntat för verkliga ferryl-intermediärer, vilket förstärker idén att den oväntat långa bindningen måste bero på mer subtila elektroniska effekter snarare än en enkel förändring i kemisk form.
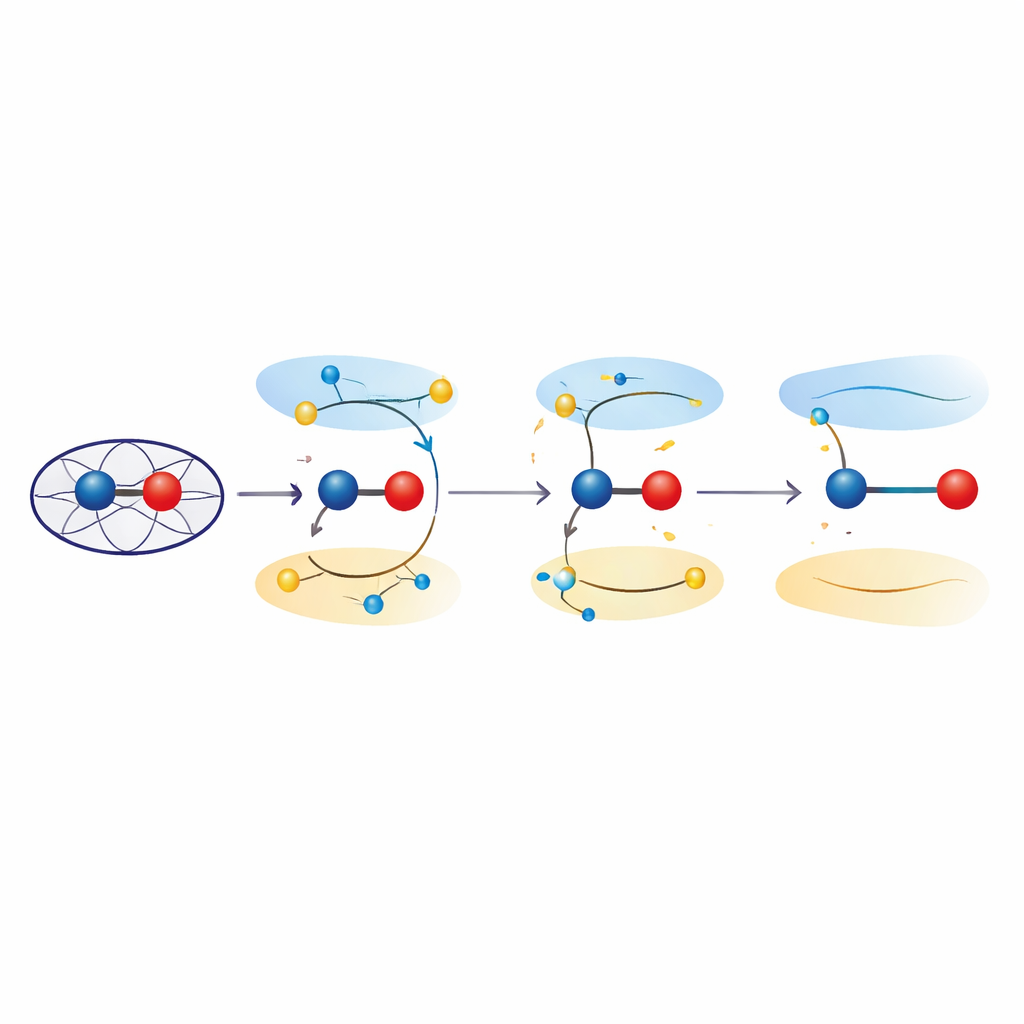
Exciterade tillstånd som töjer bindningar
För att undersöka dessa effekter använde forskarna kvantmekaniska beräkningar både på förenklade modeller och på hela proteinmiljön. Med tidsberoende täthetsfunktionalteori och kombinerade kvantmekanik/molekylmekanik-ansatser undersökte de hur främjande av elektroner från bindande till antibindande orbitaler i järn–syre-enheten ändrar den föredragna bindningslängden. Dessa exciterade tillstånd, som ligger nära grundtillståndet för ferryl i energi, gav konsekvent järn–syre-avstånd i intervallet 1,8–1,9 ångström — vilket överensstämmer med kristallografiska observationer. Analys av elektrondistributionen visade att i dessa tillstånd beter sig inte längre järn–syre-paret som en ren Fe(IV)=O-dubbelbindning, utan antar istället "ferric–oxyl"-karaktär, med egenskaper liknande Fe(III) bundet till en syrecentrerad radikal. Kvantfinslipning av de experimentella strukturerna bekräftade att sådana exciterad-tillståndsbeskrivningar passar data åtminstone lika väl som konventionella grundtillståndsmodeller.
Vad detta betyder för förståelsen av enzymkraft
Kort sagt föreslår arbetet att långa järn–syrebindningar observerade i dessa heme-peroxidaser inte kräver att man åberopar skada, reduktion eller dolda protoner. Istället kan de uppstå naturligt när ferryl-enheten kortvarigt når lågt liggande exciterade tillstånd som försvagar bindningen och ger ferric–oxyl-karaktär. För icke-specialister innebär detta att den "verkliga aktiva delen" hos många syreaktiverande enzymer kan vara mer dynamisk och elektroniskt flexibel än man tidigare trott, där subtila förskjutningar i elektronfördelningen ändrar bindningsstyrka och reaktivitet utan att förändra den övergripande kemin. Att känna igen dessa exciterade tillstånd kan förändra hur forskare tolkar strukturella data om kraftfulla biologiska oxidationsmedel och kan vägleda designen av konstgjorda katalysatorer som imiterar, eller avsiktligt finställer, denna känsliga elektroniska balansakt.
Citering: Williams, L.J., Kamps, J.J., Brânzanic, A.M.V. et al. Can ferric-oxyl excited states explain elongated iron-oxygen bonds in heme peroxidase catalytic intermediates?. Nat Commun 17, 2324 (2026). https://doi.org/10.1038/s41467-026-69192-8
Nyckelord: heme-peroxidas, ferryl-intermediär, järn-syrebindning, exciterade elektroniska tillstånd, röntgenfria-elektronlaser