Clear Sky Science · sv
En ekvivariant förtränad transformer för enhetlig 3D-representation av molekyler
Att lära datorer att se molekyler i 3D
Att designa nya läkemedel och material kräver att man förstår hur molekyler verkligen ser ut och rör sig i tre dimensioner, inte bara som platta formler på papper. Denna artikel presenterar en kraftfull ny artificiell intelligensmodell som kan lära sig av 3D-formerna hos många olika typer av molekyler samtidigt — från små läkemedelslika föreningar till stora proteiner och deras komplex — och sedan använda den kunskapen för att förutsäga hur starkt de interagerar och vilka som kan bli framtida läkemedel.
En enda karta för många molekylära världar
De flesta nuvarande AI-verktyg för kemi är specialister: en tränas bara på små molekyler, en annan bara på proteiner och en tredje bara på deras komplex. Denna uppdelning slösar med data och gör det svårt att överföra vad som lärts i ett område till ett annat. Författarna bygger istället en enda ”grundmodell”, kallad Equivariant Pretrained Transformer (EPT), som lär sig från en omfattande samling 3D-molekylstrukturer hämtade från flera offentliga databaser. Genom att behandla alla dessa strukturer inom ett gemensamt ramverk kan modellen känna igen gemensamma mönster i hur atomer ordnar sig och interagerar, oavsett om de tillhör en enkel läkemedelsmolekyl eller ett komplext nätverk av proteinkedjor. 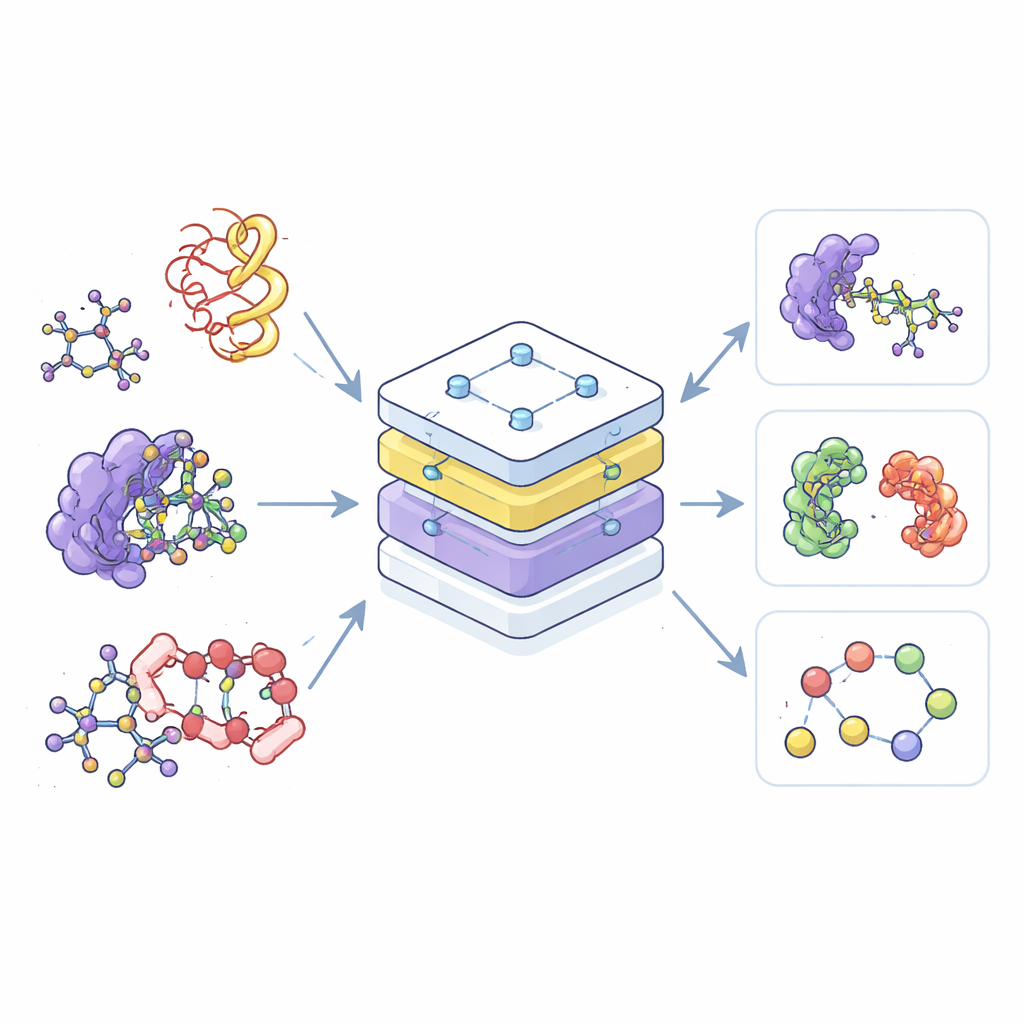
Bryta ner molekyler i hanterbara delar
För att hantera den stora variationen och storleken hos molekylära system introducerar forskarna idén om ”block” — små, meningsfulla atomsegment. För små molekyler grupperar ett block en tung atom med dess bundna väten; för proteiner blir varje aminosyra ett block. Under träningen ser modellen både de finmaskiga atomerna och den grövre blockstrukturen, vilket gör att den kan koppla lokala kemiska detaljer till bredare 3D-former som proteinryggraden eller bindningsfickor. Detta blockperspektiv skapar också ett gemensamt språk som fungerar över mycket olika molekyltyper, vilket gör det möjligt för en modell att förstå dem alla.
Lära genom att städa upp bullriga strukturer
I stället för att få explicita etiketter som ”denna molekyl är löslig” eller ”den här binder hårt” tränas EPT på ett självövervakat sätt. Författarna stör medvetet varje molekylblock genom att slumpmässigt förskjuta och rotera det bort från dess sanna position, och ber sedan modellen att härleda de krafter och vridningar som krävs för att återställa den ursprungliga strukturen. Eftersom träningen respekterar grundläggande geometriska regler — molekylen ska se likadan ut om hela systemet roteras eller flyttas i rymden — lär sig modellen en fysikaliskt rimlig känsla för 3D-form. Detta avbrusningsspel lär EPT hur atomer inom och mellan block håller ihop och hur subtila geometriska förändringar påverkar stabiliteten. 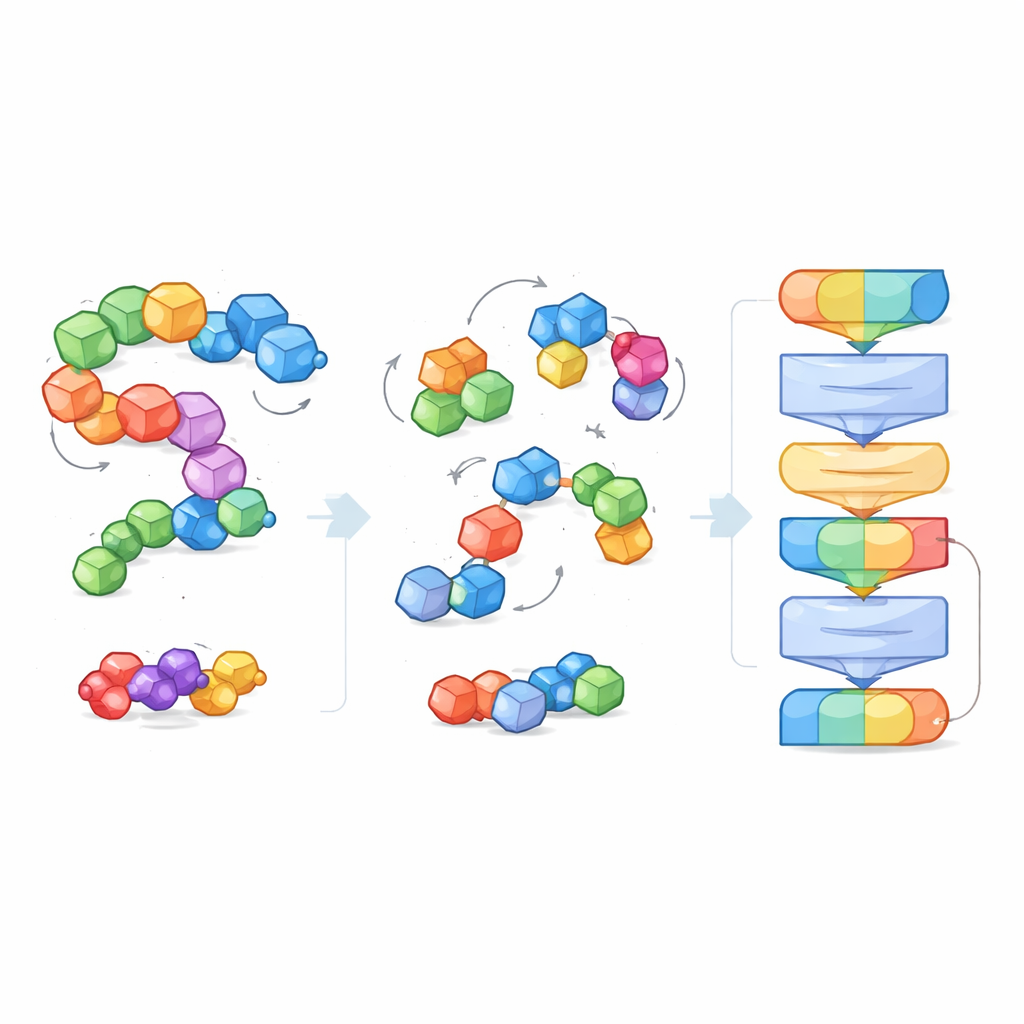
Sätta modellen på prov
Efter förträning på mer än fem miljoner strukturer finjusteras EPT för flera verkliga vetenskapliga uppgifter. Den förutsäger hur starkt en liten molekyl binder till en proteinficka, hur en enda mutation vid ett proteingränssnitt påverkar bindningen, och viktiga fysikaliska egenskaper hos små molekyler som kemister bryr sig om. Över olika benchmarks matchar eller överträffar den enhetliga modellen de bästa befintliga verktygen, inklusive sådana som varit noggrant anpassade för endast ett domän. Särskilt när den tränats på en typ av data, såsom små molekyler, hjälper den fortfarande med till synes olika uppgifter som proteinbindning — vilket visar att den fångat brett användbara kemiska principer snarare än snäva knep.
Söka efter nya COVID‑19‑behandlingar
Författarna visar ytterligare EPT:s praktiska värde genom att använda den i en omställningsutmaning för läkemedel. De finjusterar först modellen på protein–ligandkomplex och använder den sedan för att rangordna nästan 2 000 redan godkända läkemedel efter deras förutsagda förmåga att binda SARS‑CoV‑2:s huvudproteas, ett nyckelenzym som viruset behöver för att replikera. Kända anti‑COVID‑19‑läkemedel klättrar mot toppen av rankningen, och modellen lyfter fram ytterligare lovande kandidater. Tolv högst rankade molekyler granskas närmare med datorbaserade simuleringar, och två — inklusive en som ursprungligen inte utvecklades för COVID‑19 — visar särskilt stark förutsagd bindning och bekräftas experimentellt att hämma det virala proteaset på mikromolära nivåer.
Ett steg mot generell molekylär AI
Enkelt uttryckt visar detta arbete att en enda, geometri‑medveten AI‑modell kan lära sig en gemensam 3D‑förståelse av många molekylära system och sedan använda den för att besvara en rad vetenskapliga frågor. Genom att organisera molekyler i block och träna modellen att ”laga” förvrängda strukturer skapar författarna ett verktyg som inte bara förutsäger siffror mer exakt utan också kan påskynda uppgifter som att hitta nya antivirala läkemedel. EPT pekar mot en framtid där allmänna molekylära AI‑system hjälper kemister och biologer att utforska kemiskt rum mer effektivt, styra experiment och förkorta vägen från atomstruktur till praktiska terapier och material.
Citering: Jiao, R., Kong, X., Zhang, L. et al. An equivariant pretrained transformer for unified 3D molecular representation learning. Nat Commun 17, 2606 (2026). https://doi.org/10.1038/s41467-026-69185-7
Nyckelord: 3D-molekylrepresentation, ekvivariant transformer, Läkemedelsupptäckt, protein–ligand-bindning, självövervakad inlärning