Clear Sky Science · sv
Ensembles med atomupplösning av intrinsiskt oordnade proteiner med AlphaFold
Varför formskiftande proteiner är viktiga
Våra celler är fyllda av proteiner som aldrig lägger sig till ro i en enda, stel form. Dessa ”intrinsiskt oordnade” proteiner beter sig mer som sladdriga nudlar än prydligt hopvikta maskiner, men de är centrala för processer som spänner från cellsignalering till neurodegenerativa sjukdomar. Eftersom de ständigt rör sig och böjer sig är det extremt svårt att fånga hela deras formregister med atomdetalj, och det kräver vanligtvis år av experiment och beräkningar. Denna artikel presenterar ett nytt sätt att kombinera artificiell intelligens och fysik för att kartlägga dessa rastlösa molekyler mycket effektivare.
Utmaningen med rastlösa molekyler
Olika från lärobokens proteinmodeller som visar en prydlig struktur, vandrar intrinsiskt oordnade proteiner (IDP:er) genom ett stort landskap av möjliga former. Den flexibiliteten hjälper dem att känna igen många olika partners, men gör dem också notoriskt svåra att studera. Traditionella laboratorietechniker, som avancerad kärnmagnetisk resonans och röntgenspridning, kan ge genomsnitt över många former men inte varje enskild konformation. Datorsimuleringar med full atomdetalj kan i princip följa varje atom när ett IDP slingrar sig, men de är mycket kostsamma och beroende av fint inställda fysikaliska modeller. Som en följd har forskarsamhället endast en begränsad samling av noggranna, detaljerade IDP-ensembler att lära av.
Kombinera smarta gissningar med fysikaliska regler
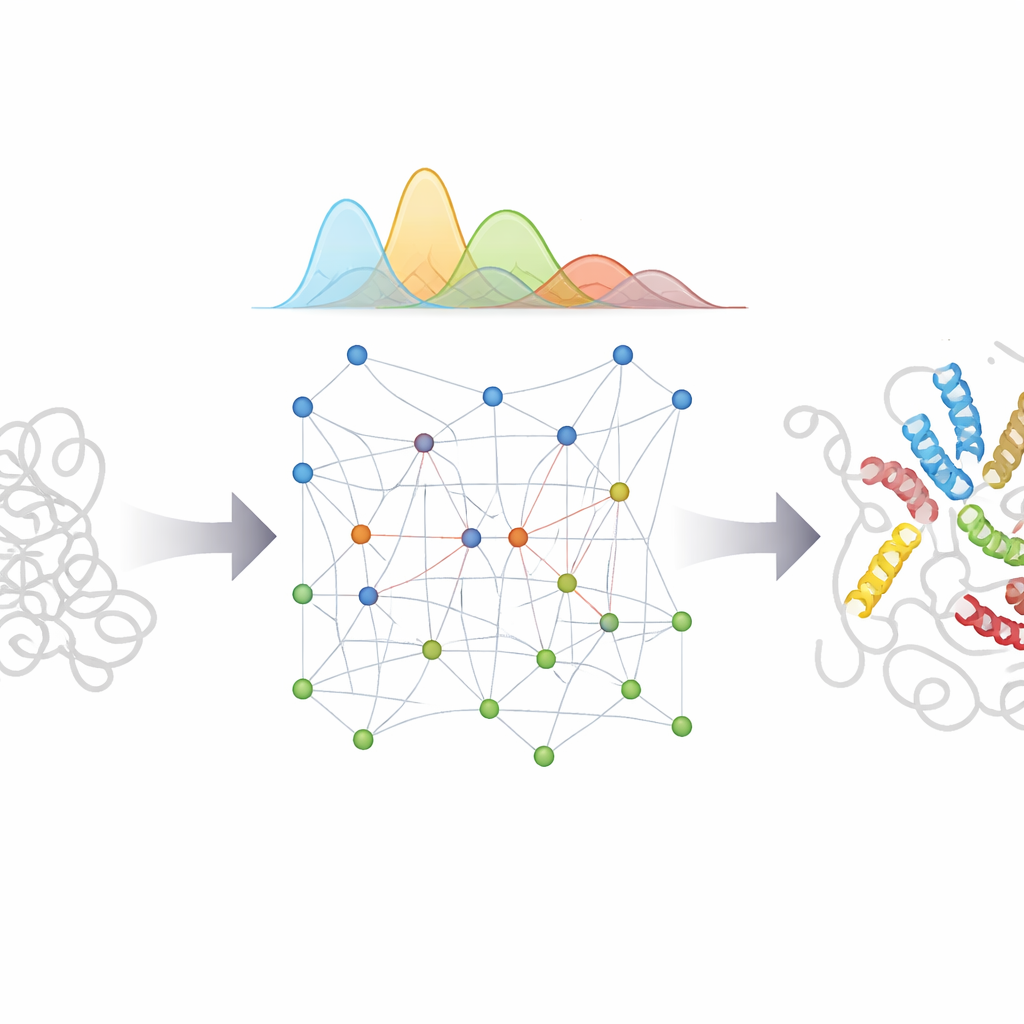
Under de senaste åren har AlphaFold-familjen av djupinlärningsverktyg förbluffat biologin genom att förutsäga proteinstrukturer från deras aminosyrasekvenser. För oordnade proteiner är dock AlphaFolds vanliga styrka—att gissa en enda bästa form—mindre användbar, eftersom IDP:er inte har bara en. Det AlphaFold däremot ger är rik information om hur sannolikt det är att olika delar av kedjan är nära eller långt ifrån varandra. Författarna byggde ett nytt ramverk, kallat bAIes, som behandlar denna AI- härledda information som mjuk vägledning och blandar den med en snabb, fysikbaserad modell som medvetet utgår från en ”slumpmässig coil”-syn, där kedjan utforskar alla möjliga böjningar och vridningar utan att favorisera någon särskild struktur.
Från slumpmässiga trassel till realistiska ensembler
Först konstruerade forskarna en effektiv fysikalisk modell som reproducerar hur en helt ostrukturerad proteinkedja beter sig, baserat på statistik som extraherats från tusentals kända proteinstrukturer. Denna modell tjänar som ”prior”—den grundläggande förväntningen om hur ett IDP rör sig om vi inte vet något annat. Nästa steg är att bAIes läser AlphaFolds prediktioner om vilka restpar som tenderar att komma nära varandra. Istället för att tvinga proteinet in i ett enda mönster omvandlar det dessa antydningar till milda distansrestriktioner med inbyggd osäkerhet, vilket tillåter kedjan att endast uppfylla AIs förslag när de är förenliga med den bredare fysikaliska bilden.
Testning mot verkliga experiment
För att se om detta angreppssätt fungerar applicerade gruppen bAIes på en uppsättning av 21 proteiner som sträckte sig från nästan fullständiga slumpmässiga coilar till mer komplexa system med övergående helixar och flera domäner. För varje protein jämförde de de datorgenererade ensemblerna med en mängd experimentella mätningar som undersöker både lokala detaljer och global storlek och form. För mycket sladdriga proteiner, såsom det Alzheimers-relaterade peptiden Aβ40, var den enkla slumpcoilmodellen redan nära verkligheten, och bAIes bevarade denna goda överensstämmelse. För delvis strukturerade proteiner förbättrade bAIes överensstämmelsen med experimenten genom att korrekt fånga var korta helixsegment och kompakta fläckar uppträder och försvinner. Avgörande är att metoden förblev robust även när AlphaFold var överdrivet självsäker och felaktigt förutsade stabila veck där lösningsexperiment visar oordning, eftersom bAIes uttryckligen tillåter fel i AI-inputen.
Slår eller matchar befintliga metoder
Författarna ställde sedan bAIes mot tunga all-atom-simuleringar körda på specialiserade superdatorer, ledande grovskaliga modeller som förenklar proteiner till kulor, och nya djupinlärningsgeneratorer tränade på simuleringsdata. I flera tester matchade eller överträffade bAIes konsekvent dessa angreppssätt när det gäller att reproducera experimentella data, samtidigt som det krävde mycket mindre beräkningsresurser än fullskaliga simuleringar. Det fungerade också utanför enkla IDP:er, och hanterade proteiner med flera styva domäner länkade av flexibla kopplingar och återvann deras övergripande former i lösning. När forskarna dessutom finjusterade bAIes-ensemblerna med experimentella data förbättrades överensstämmelsen ytterligare, vilket visar att metoden kan fungera som en kraftfull utgångspunkt för integrativ modellering.
Vad detta betyder för biologi och medicin
Genom att förena AlphaFolds mönsterigenkänningskraft med en omsorgsfullt utformad fysikalisk modell och en bayesiansk behandling av osäkerhet erbjuder bAIes en praktisk väg till detaljerade ”filmer” av oordnade proteiner snarare än enstaka stillbilder. Dessa atomärt detaljerade ensembler kan hjälpa forskare förstå hur flexibla regioner känner igen partners, hur felveckning och aggregering börjar i sjukdomar som Parkinsons och Alzheimers, och hur små molekyler kan binda till svårfångade, skiftande mål. Eftersom metoden är effektiv och byggd i öppen källkod kan den brett antas för att generera realistiska ensembler för många oordnade proteiner, vägleda experiment och stödja framtida AI-system som syftar till att förutsäga inte bara en struktur utan hela spektrat av former som livets mest flexibla molekyler kan anta.
Citering: Schnapka, V., Morozova, T.I., Sen, S. et al. Atomic resolution ensembles of intrinsically disordered proteins with Alphafold. Nat Commun 17, 2399 (2026). https://doi.org/10.1038/s41467-026-69172-y
Nyckelord: intrinsiskt oordnade proteiner, AlphaFold, Bayesiansk modellering, proteinensembler, strukturell biologi