Clear Sky Science · sv
Prediktion av kemisk likhet mellan tandemmasspektra i olika joniseringslägen inom metabolomik
Varför det är viktigt att koppla kemiska punkter
Varje klunk kaffe, andetag eller läkemedelskur lämnar små kemiska spår i våra kroppar. Moderna instrument kan upptäcka tusentals av dessa molekyler samtidigt, men att översätta signalerna till biologisk förståelse är fortfarande förvånansvärt svårt. Denna studie presenterar MS2DeepScore 2.0, ett maskininlärningsverktyg som hjälper forskare att se hur dessa molekyler hänger ihop, även när signalerna registreras på mycket olika sätt. Genom det lovar metoden snabbare och mer fullständiga tolkningar av komplexa kemiska blandningar inom medicin, näringslära och miljöforskning.
Två sätt att betrakta samma molekyl
Masspektrometri är en grundläggande teknik som väger och sönderdelar molekyler för att avslöja deras identitet. I rutinexperiment mäter forskare ofta samma prov i två lägen: ett som gynnar positivt laddade joner och ett som gynnar negativt laddade joner. Varje läge ger sitt karakteristiska "streckkod" av fragment. Även när båda mätningarna kommer från samma molekyl kan de resulterande mönstren se så olika ut att traditionella jämförelsemetoder misslyckas. Som en följd analyserar forskare vanligtvis de två lägena separat, bygger två oberoende kartor av provet och riskerar att missa viktiga relationer mellan kemikalier.
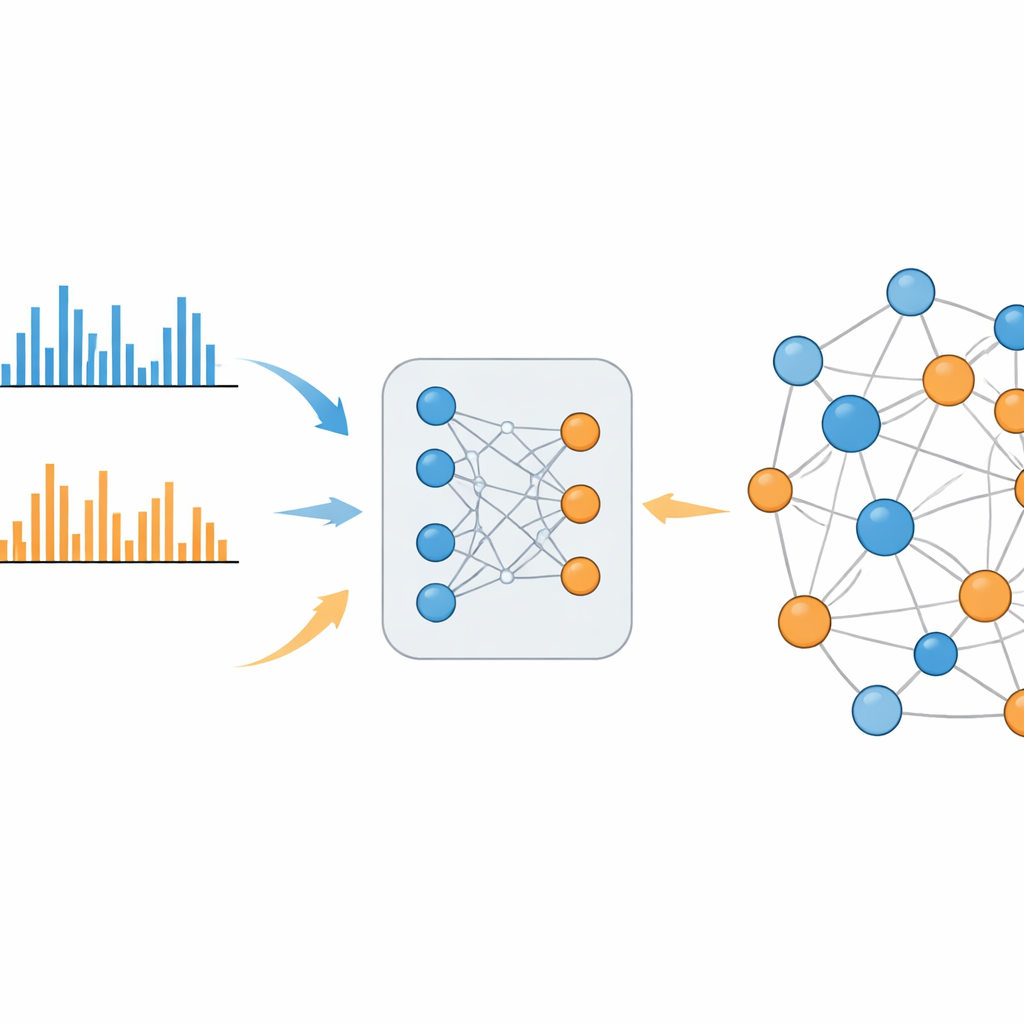
En lärande metod som överbryggar klyftan
MS2DeepScore 2.0 tar sig an denna klyfta genom att lära sig kemisk likhet direkt från stora bibliotek av kända spektra. Modellen bygger på en tvilling-neuralt nätverksarkitektur som omvandlar varje fragmenteringsmönster till ett 500-nummerlångt fingeravtryck, kallat en embedding. Under träningen ser systemet hundratusentals exempel från både positivt och negativt läge, tillsammans med hur lika de bakomliggande molekylerna faktiskt är. Det justerar sig så att spektra från besläktade molekyler får liknande embeddingar, oavsett om de mättes i samma eller motsatta lägen. Den nya versionen går längre än sin föregångare genom att mata in extra information, som massan av ursprungsmolekylen och vilket joniseringsläge som användes, samt genom att använda ett omsorgsfullt balanserat urvalsschema så att sällsynta men informativa kemiska relationer inte drunknar i vanliga, icke-informativa sådana.
Från spridda signaler till enade kartor
När modellen är tränad kan MS2DeepScore 2.0 uppskatta hur kemiskt lika två spektra är, inklusive par mellan positivt och negativt läge. Författarna visar att dessa prediktioner korrelerar väl med etablerade mått på strukturell likhet, inte bara inom varje läge utan även över lägen. Med verkliga data från mänsklig urin, mänskligt blodplasma och en vild ätbar växt bygger de "molekylära nätverk" där varje spektrum är en nod och stark predikterad likhet skapar en förbindelse. Till skillnad från äldre tillvägagångssätt blandar dessa nätverk naturligt data från positivt och negativt läge till en enda, sammanhängande karta. Klastrar kuraterade av experter inkluderar till exempel grupper av koffeinrelaterade molekyler i urin som är förbundna över joniseringslägen och stämmer överens med kända metaboliska vägar.
Se det kemiska landskapet på en blick
Molekylära nätverk är kraftfulla men kan bli trassliga om för många svaga länkar inkluderas. För att undvika detta använder författarna MS2DeepScore:s embeddingar direkt som koordinater i ett tvådimensionellt upplägg skapat med en teknik kallad UMAP. Varje punkt i denna karta representerar ett spektrum, och närliggande punkter motsvarar molekyler som modellen anser vara kemiskt lika. Positiva och negativa läges-spektra av samma förening, som vid en första anblick ser mycket olika ut, hamnar ofta sida vid sida i detta embeddingsutrymme. Teamet tränar också en extra modell som granskar varje embedding och uppskattar hur tillförlitlig den är, och flaggar spektra som är brusiga, ofullständiga eller olik allt som setts under träning. Genom att ta bort dessa lågkvalitetspunkter förbättras den övergripande noggrannheten och visualiseringarna blir mer trovärdiga.
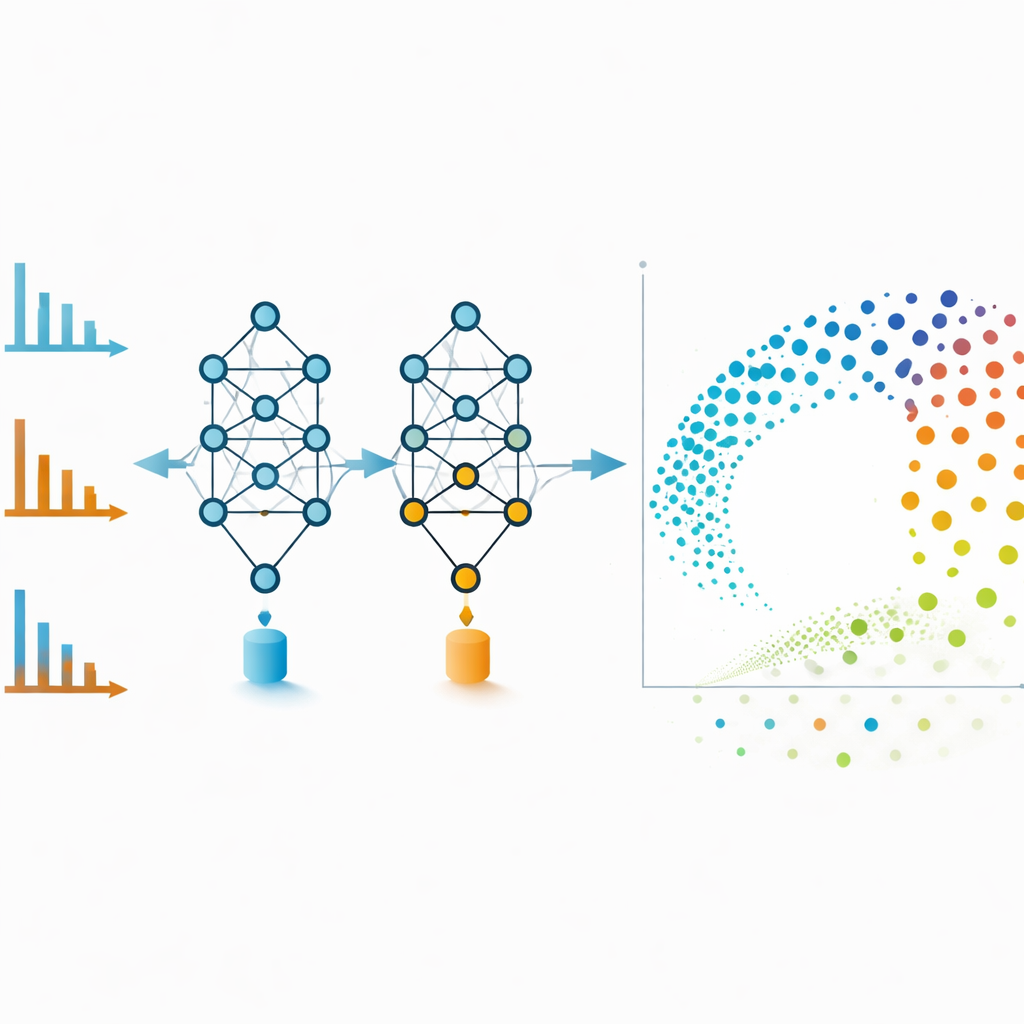
Göra avancerade verktyg tillgängliga i vardagslaboratorier
För att säkerställa att denna teknik kan användas även av andra än programmeringsspecialister har författarna integrerat MS2DeepScore 2.0 i populär, fritt tillgänglig masspektrometris programvara. Med denna integration kan forskare upptäcka signaler, bygga molekylära nätverk som ignorerar gränser mellan joniseringslägen och utforska det resulterande kemiska rummet via interaktiva instrumentpaneler. Koden, tränade modeller och exempel-dataset delas öppet, och systemet kan tränas om eller finjusteras för specialiserade kemiska klasser.
Vad detta betyder för framtida upptäckter
För icke-specialister är huvudbudskapet att MS2DeepScore 2.0 hjälper till att omvandla fragmenterade och lägesberoende mätningar till en enda, mer begriplig bild av vilka molekyler som finns i ett prov. Genom att pålitligt länka signaler som tidigare levde i separata analytiska världar låter metoden forskare utnyttja mycket större referensbibliotek, jämföra prover mer fullständigt och rikta uppmärksamheten mot meningsfulla kluster av besläktade föreningar. Denna korskoppling av data förväntas påskynda identifieringen av biomarkörer, näringsämnen, naturliga produkter och föroreningar, och i förlängningen fördjupa vår förståelse av hur kemi påverkar hälsa och miljö.
Citering: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Nyckelord: metabolomik, masspektrometri, maskininlärning, molekylärt nätverkande, kemisk likhet