Clear Sky Science · sv
Modellering av lungsjukdom med humana iPSC visar att hämning av p300/CBP undertrycker alveolärt övergångstillstånd
Varför ärrbildning i lungorna berör oss alla
Idiopatisk lungfibros (IPF) är en obeveklig sjukdom där lungvävnad gradvis omvandlas till ärrvävnad, vilket gör varje andetag tyngre. Dagens läkemedel kan endast bromsa denna process och ger ofta besvärande biverkningar. I denna studie används avancerade stamcells‑ och genomiska verktyg för att återskapa ärrade lungor i labbet och ställa en enkel men avgörande fråga: kan vi hitta en brytare som styr skadade lungceller bort från ett skadligt tillstånd och tillbaka mot läkning?
Ett labbodlat fönster mot en ärrad lunga
För att studera IPF byggde forskarna miniaturiserade lungor från humana inducerade pluripotenta stamceller (iPSC). Dessa iPSC styrdes att bli alveolära celler — cellerna som bekläder de små luftblåsorna där syre går över till blodet — och odlades tillsammans med lungfibroblaster, stödjeceller som bildar ärr. Bäddade i ett mjukt gelmedium uppvisade dessa ”alveolära organoider” beteenden som påminde om verklig lungvävnad. När de exponerades för cytostatikumet bleomycin, en känd utlösare av lungskada, krympte gelen när fibroblasterna drog i den, vilket efterliknade den vävnadskontraktion som ses vid fibros.
Med detta system screenade teamet ett bibliotek med 264 små molekyler och mätte automatiskt hur mycket varje läkemedel förhindrade gelkontraktion, med ett djupinlärningsbaserat bildanalysverktyg för objektiva avläsningar. Många föreningar hade ingen effekt, men en familj utmärkte sig klart: hämmare av proteinerna p300 och CBP, som hjälper till att styra hur DNA packas och vilka gener som aktiveras. Alla åtta p300/CBP‑inriktade föreningar i biblioteket minskade kontraktionen vid låga doser, vilket pekar ut denna väg som ett lovande grepp på fibros. 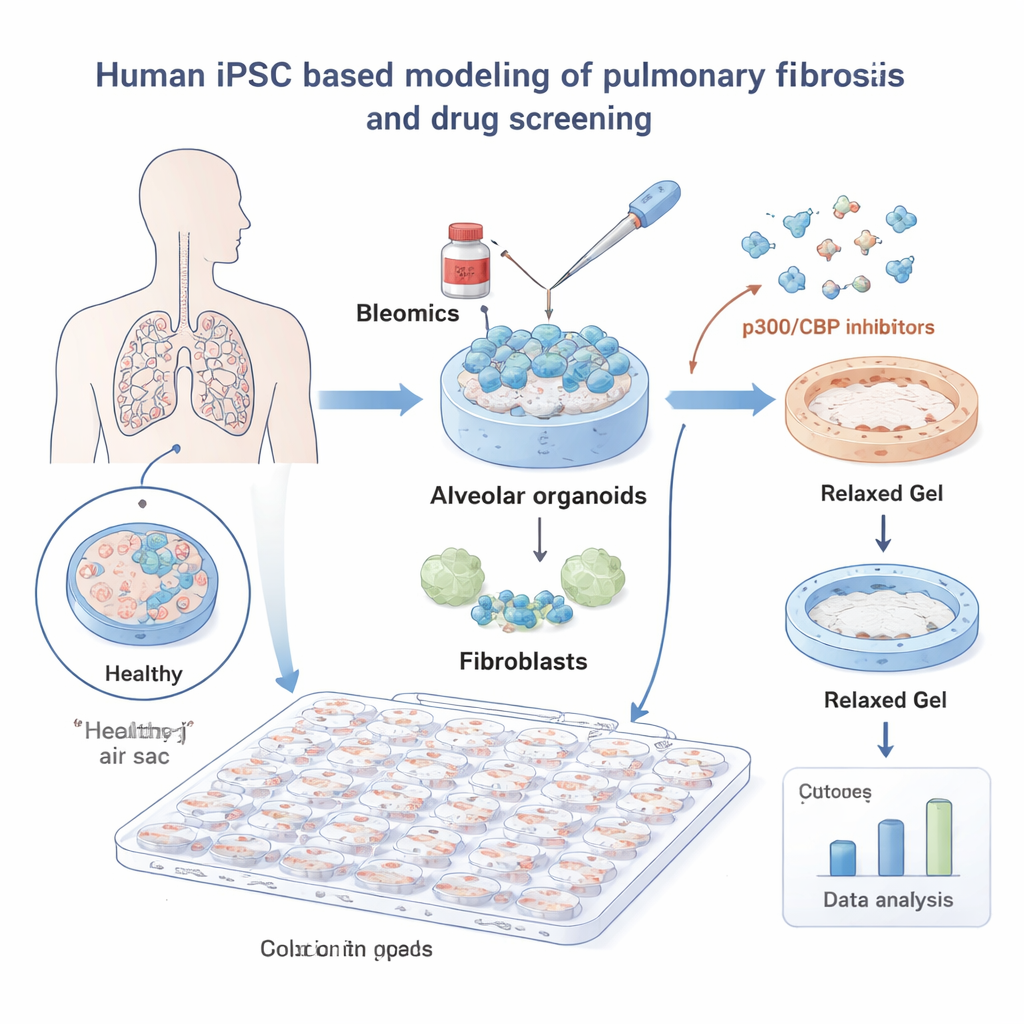
Problemorsakarna: övergångslungceller
Nyare forskning har identifierat en problematisk ”mellanliggande” celltyp i sjuka lungor, kallad det alveolära övergångstillståndet. Normalt mognar stödjeceller kända som AT2‑celler till ultratunna AT1‑celler som täcker alveolerna och möjliggör gasutbyte. Vid IPF stannar dock ofta AT2‑celler kvar i detta övergångstillstånd, uttrycker stress‑ och reparationsgener samtidigt som de misslyckas med att slutföra övergången till fullt fungerande AT1‑celler. Dessa övergångsceller klustrar sig i fibrotiska områden och kommunicerar starkt med fibroblaster, men det har varit oklart om de bara är en följd av skadan eller aktiva drivkrafter bakom ärrbildning.
Genom att sekvensera RNA och profilera öppet kromatin i sina organoider visade författarna att de övergångsceller som uppstod i deras modell nära motsvarade dem som finns i lungor från IPF‑patienter. Dessa inducerade övergångsceller visade gensignaturer för stress, inflammation och matrixombyggnad, och de aktiverade kraftigt de medodlade vuxna lungfibroblasterna. Avgörande var att när p300/CBP blockerades sjönk markörer för övergångstillståndet, AT2‑identiteten bevarades bättre och fibroblastaktiveringen avtog. Med andra ord förgiftade inte läkemedlen cellerna i allmänhet; de förhindrade selektivt att AT2‑celler fastnade i detta skadliga limbo.
Att nysta upp de molekylära brytarna
För att förstå hur p300/CBP formar detta ödesval undersökte teamet kemiska markörer på histoner — proteiner som hjälper till att paketera DNA. En särskild markör, H3K27‑acetylering, läggs ofta ned av p300/CBP vid aktiva förstärkare och promotorer. I övergångsceller bar regioner nära stress‑respons‑ och profibrotiska gener stark H3K27‑acetylering och var berikade för bindningsställen för transkriptionsfaktorer som AP‑1 och HNF1B. När celler behandlades med p300/CBP‑hämmare minskade dessa acetylmärken vid dessa platser och uttrycket av många profibrotiska gener föll. Att blockera AP‑1 direkt, eller reducera AP‑1 och HNF1B med små interfererande RNA, dämpade också övergångsprogrammet och organoidkontraktionen, vilket knöt denna trio — p300/CBP, AP‑1 och HNF1B — till motorn som driver fibrotisk ombyggnad.
Bortom petriskålen testade studien en hämmare, CBP30, i möss med bleomycininducerad lungskada. Djur som gavs CBP30 hade färre övergångsepitelceller, mindre aktivering av ärrbildande myofibroblaster och minskat uttryck av fibrosmarkörer. Denna korsvalidering mellan humana stamcellsmodeller och en djurmodell stärker argumentet att p300/CBP inte bara är ett laboratoriefenomen utan en verklig regulator av lungsårbildning. 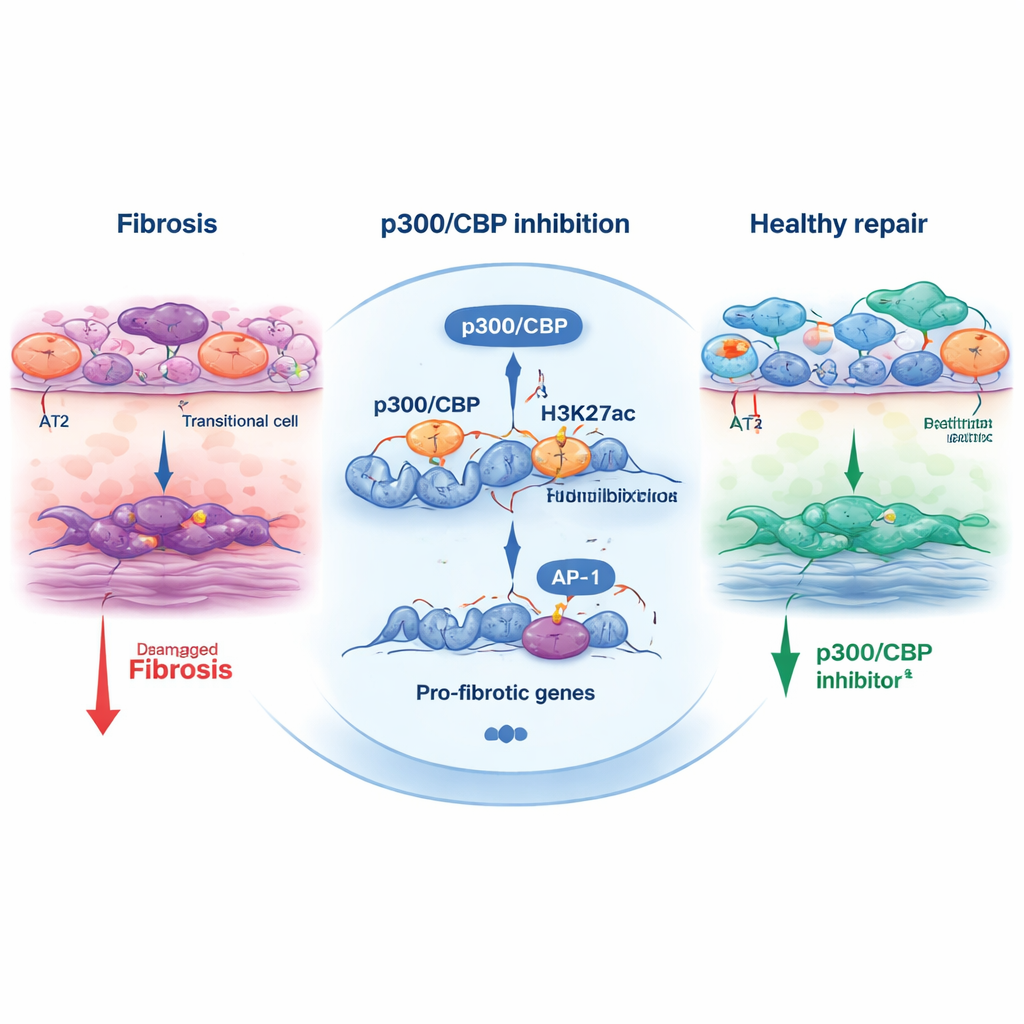
Vad detta betyder för framtida behandlingar
För icke‑specialister är huvudpoängen att författarna byggt en realistisk human modell av fibrotiska lungor och använt den för att lyfta fram ett nytt läkemedelsmål. Deras arbete tyder på att lungsårbildning delvis drivs av ett reversibelt, stressinducerat övergångstillstånd som leder den omgivande vävnaden på villovägar. Genom att dämpa p300/CBP kan det bli möjligt att tysta detta tillstånd, hålla alveolära celler på en hälsosam utvecklingsbana och minska signalerna som puttar fibroblaster i övervarv. Även om p300/CBP‑hämmare fortfarande behöver optimeras för säkerhet och prövas kliniskt, pekar denna studie mot behandlingar som angriper den grundläggande cellulära misskommunikationen i IPF istället för att enbart bromsa dess följder.
Citering: Tsutsui, Y., Masui, A., Konishi, S. et al. Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State. Nat Commun 17, 1214 (2026). https://doi.org/10.1038/s41467-026-68909-z
Nyckelord: idiopatisk lungfibros, alveolära organoider, p300/CBP‑hämmare, övergångsepitelceller, lungstamceller