Clear Sky Science · sv
Pålitlig förutsägelse av Enzyme Commission‑nummer med en hierarkisk tolkbar transformer
Varför det är viktigt att förutsäga enzymers uppgifter
Varje levande cell drivs av otaliga små kemiska maskiner som kallas enzymer. Varje enzym har en specifik “uppgift”, och den uppgiften kodas i ett Enzyme Commission (EC)-nummer, en fyradelad kod lite som en postadress. Att korrekt tilldela EC‑nummer är avgörande för att förstå metabolism, utforma nya läkemedel, konstruera mikrober som tillverkar bränslen eller plastalternativ och följa hur ekosystem bearbetar kemikalier. Men experiment som bestämmer enzymfunktioner är långsamma och kostsamma. Denna studie presenterar HIT‑EC, en ny artificiell intelligensmodell som på ett tillförlitligt sätt kan förutsäga EC‑nummer från proteinsekvenser samtidigt som den förklarar varför den gjorde varje förutsägelse.
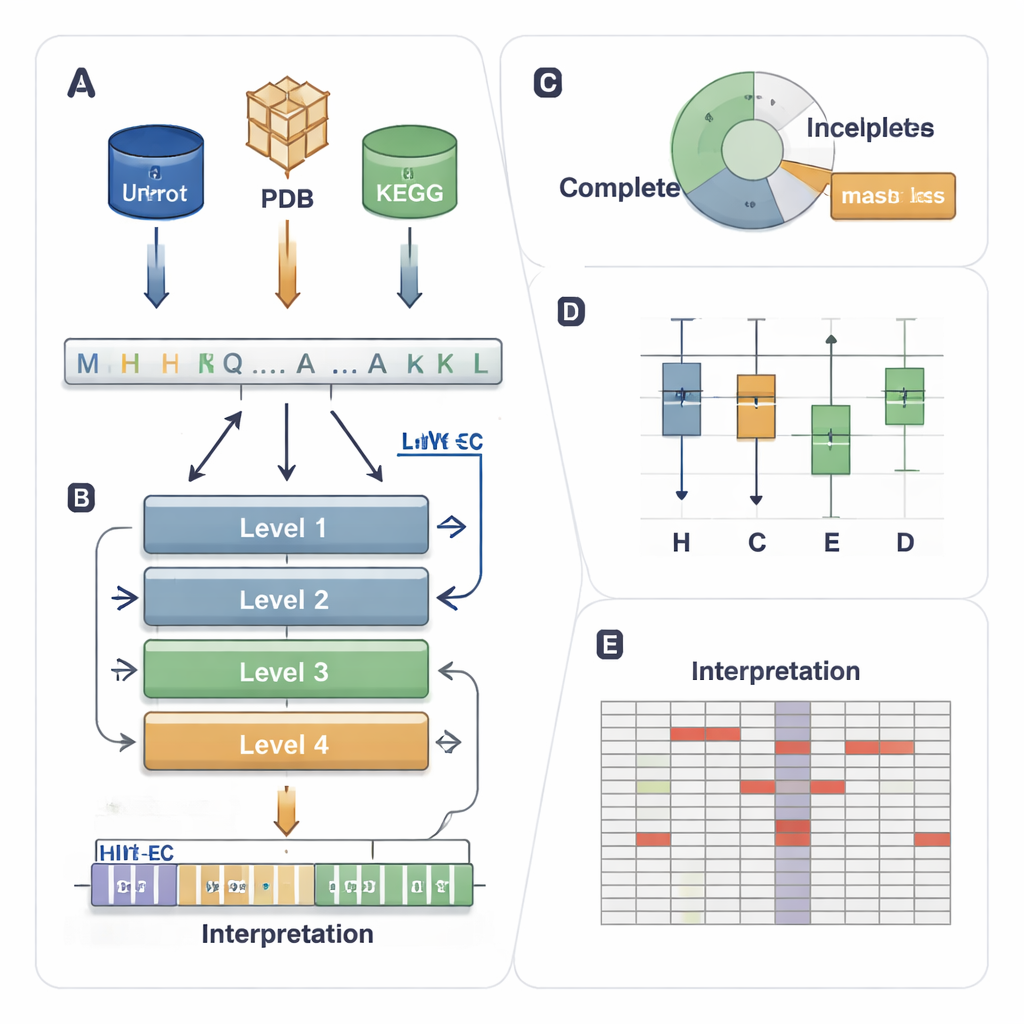
En postadresseringsmetod för enzymuppgifter
EC‑systemet tilldelar varje enzym en fyranivåkod, till exempel 1.1.1.37. Den första siffran anger en bred klass (till exempel enzymer som flyttar elektroner eller överför grupper) och de senare siffrorna beskriver mer precisa reaktionsdetaljer. Denna hierarki är kraftfull men skapar ett krävande prediktionsproblem: en modell måste få alla fyra nivåerna rätt för tusentals möjliga koder, även när vissa enzymer är sällsynta eller bara delvis annoterade i databaser (till exempel 3.5.-.-, där de detaljerade nivåerna saknas). Befintliga datormetoder använder antingen 3D‑struktur, sekvenslikhet eller djupinlärning, men de har ofta svårt med ovanliga enzymer, ignorerar delvis märkta data och fungerar vanligtvis som ”svarta lådor” som ger liten insikt i varför ett beslut fattades.
En fyravånings‑AI som följer EC‑stegen
HIT‑EC (Hierarchical Interpretable Transformer for EC prediction) är byggd för att spegla den fyrastegs EC‑hierarkin. Den tar en rå proteinsekvens och passerar den genom fyra transformerlager, där varje lager fokuserar på en EC‑nivå. Lokala flöden kopplar varje nivå till den föregående, vilket säkerställer att ett finfördelat beslut (den fjärde siffran) måste vara förenligt med bredare beslut (första och andra siffran). Parallellt håller ett globalt flöde hela sekvenskontexten synlig i varje steg. Modellen kan också tränas på sekvenser med ofullständiga etiketter genom att använda en ”masked loss” som enkelt ignorerar saknade EC‑nivåer istället för att kasta bort sekvensen. Detta låter HIT‑EC lära sig från den stora andel proteiner i välkurerade databaser som endast är delvis annoterade.
Slår konkurrenterna i noggrannhet och hastighet
Författarna sammanställde en stor, noggrant filtrerad datamängd på cirka 200 000 enzymer med 1 938 olika EC‑nummer från Swiss‑Prot och Protein Data Bank. I upprepade hold‑out‑tester slog HIT‑EC tre ledande metoder (CLEAN, ECPICK och DeepECtransformer) både när det gäller övergripande och per‑klass F1‑poäng, som mäter balansen mellan korrekta träffar och falska larm. Den var särskilt stark på underrepresenterade EC‑koder med 25 eller färre kända exempel, där tidigare metoder ofta har svårt. HIT‑EC generaliserade också väl till nya enzymer som lades till i Swiss‑Prot efter träning och till hela genom från olika bakterier, inklusive välstuderade stammar av Escherichia coli, Bacillus subtilis och Mycobacterium tuberculosis. Trots sin sofistikering var modellen mycket effektiv: på en standard‑GPU bearbetade den ett protein på cirka 38 millisekunder—tio‑tals gånger snabbare än vissa konkurrenter som är beroende av långsammare likhetssökningar eller ensemblemetoder med många modeller.
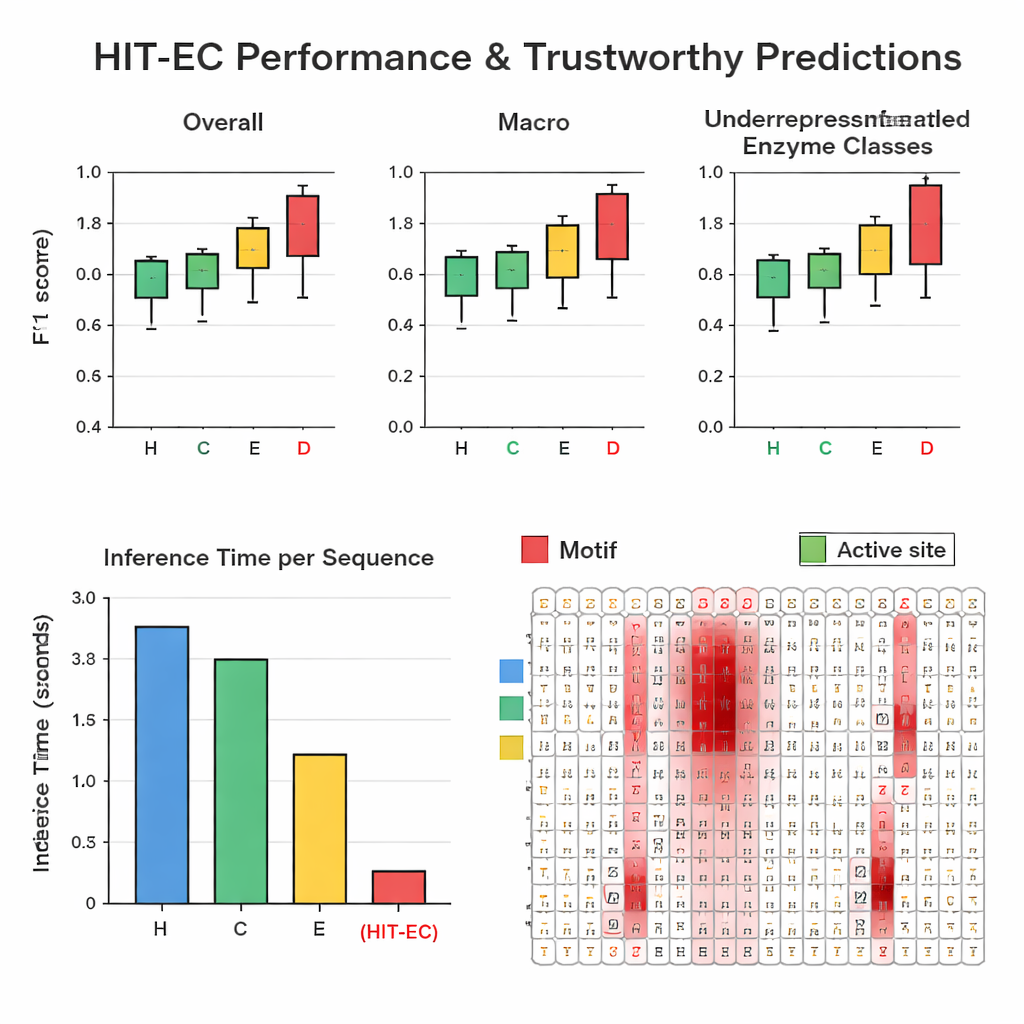
Att se vad modellen "tittar på"
För att göra sina förutsägelser tillförlitliga är HIT‑EC utformad för att visa vilka aminosyror i sekvensen som påverkade beslutet på varje EC‑nivå. Författarna byggde en tolkningsväg som kombinerar attention‑vikter med gradientinformation för att poängsätta hur viktiga varje position är. De validerade dessa poäng på väldefinierade enzymfamiljer. Till exempel i en cytochrom P450‑familj (CYP106A2) markerade HIT‑EC kända funktionella motiv såsom syrebindande och hembindande regioner, och identifierade ett subtilt EXXR‑motiv som en referensmetod missade. För klassiska representanter för varje toppnivå i EC—som alkoholdehydrogenas, hexokinas och karbanhydras—lyste modellens relevanspoäng upp läroboks‑signaturmotiv och substratbindande säten. Dessa tolkningar ger biokemiska ”bevis” för att modellen baserar sina slutsatser på meningsfulla egenskaper, inte på tillfälliga korrelationer.
Vägledning för arbete med sällsynta och nya enzymer
Gruppen testade vidare HIT‑EC på två dåligt studerade enzymer som är viktiga för föroreningar: en cytochrom P450 involverad i nedbrytning av aromatiska föroreningar, och en PET‑nedbrytande hydrolas från Streptomyces som hjälper till att bryta ner plastrelaterade molekyler. Båda enzymerna hade experimentellt karaktäriserats men saknade officiella EC‑tilldelningar. HIT‑EC förutsade korrekt de förväntade EC‑numren och framhävde motivmönster och katalytiska rester som stämmer med vad som är känt från strukturella och biokemiska studier. Sammantaget visar arbetet att HIT‑EC inte bara kan tilldela EC‑nummer mer exakt och snabbare än nuvarande verktyg, särskilt för sällsynta funktioner, utan också belysa varför ett visst enzym bedöms utföra en given kemisk uppgift. Denna kombination av prestanda och tolkbarhet gör det till en lovande motor för storskalig, pålitlig enzymannotering inom genomik, bioteknik och miljöforskning.
Citering: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Nyckelord: förutsägelse av enzymfunktion, djupinlärning i biologi, transformermodeller, proteinanmärkning, bioremediering‑enzymer