Clear Sky Science · sv
En alternativ EGFR‑aktivering genom patient‑härledd R252C‑mutation främjar cancerprogression
När cellernas antenner går på villovägar
Varför fortsätter vissa cancerformer växa trots omgångar av cytostatika och moderna immunterapier? I denna studie följer forskarna en patient med tumörer både i lungan och i hjärnan och spårar sjukdomen tillbaka till en liten förändring i en viktig cell‑yt‑"antenn" kallad EGFR. Genom att kartlägga hur just denna enda mutation omlägger tillväxtsignaler förklarar teamet inte bara patientens aggressiva cancer, utan visar också hur ett redan godkänt läkemedel, afatinib, kan tygla den.
En sällsynt mutation med stora följder
EGFR är en receptor som sitter tvärs över cellmembranet och hjälper cellen att svara på tillväxtsignaler. Många lung‑ och hjärntumörer bär på mutationer i EGFR, men de flesta kända förändringarna finns inne i cellen, i den del som fungerar som en enzymatisk omkopplare. Här upptäckte forskarna en ovanlig förändring på utsidan av EGFR, i den del som normalt binder till tillväxtfaktorer. I en patient med både lungcancer och gliom fann de att en aminosyra vid position 252 bytts från arginin till cystein — benämnt EGFR R252C. Genom att granska cancerregister identifierade de denna mutation i en liten andel gliompatienter och nästan aldrig i lungtumörer, vilket tyder på att den är sällsynt men verklig. Med hjälp av genredigering återskapade författarna exakt denna mutation i flera mänskliga hjärn‑ och lungcancercellinjer för att undersöka dess effekt.
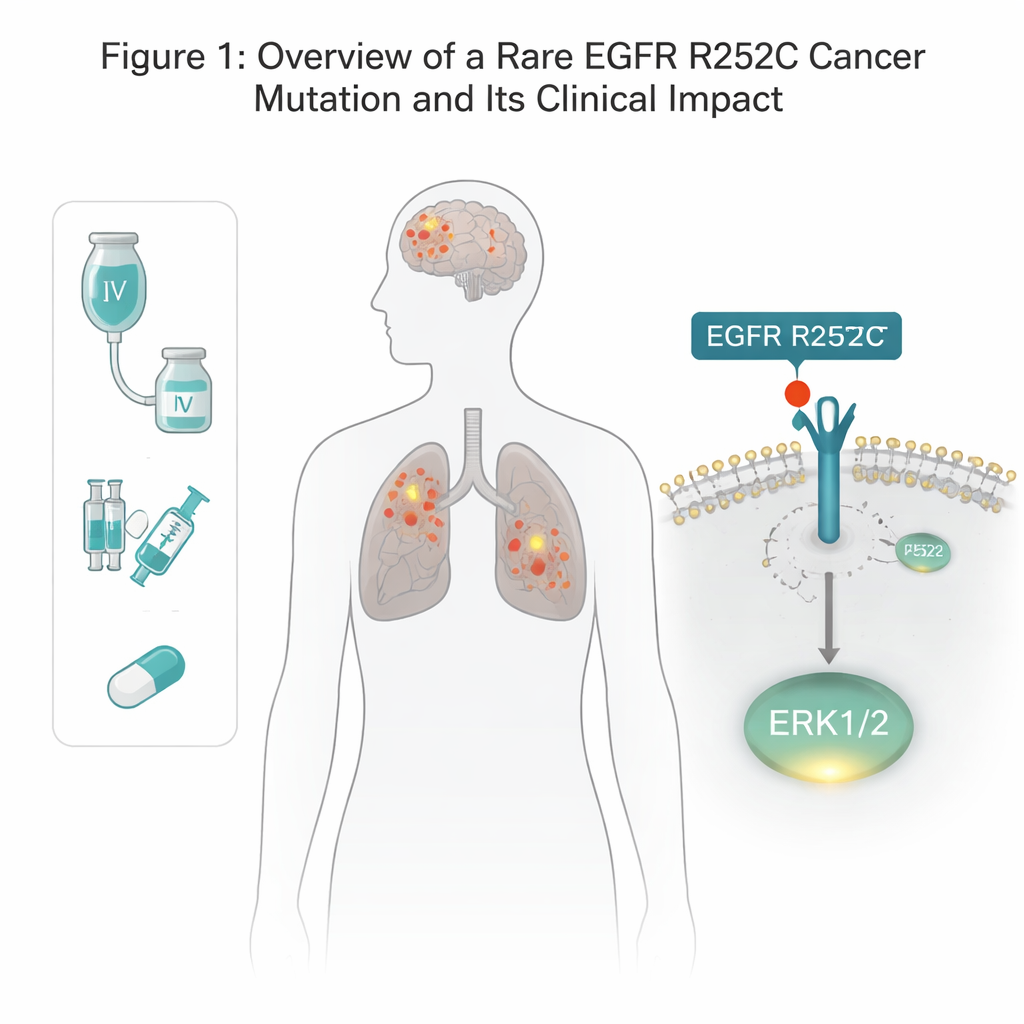
En ny genväg till tillväxtsignaler
Vanligtvis måste EGFR bilda par med en annan kopia och sedan märka sin inre svans med fosfatgrupper innan den kan slå på nedströms tillväxtvägar. Överraskande nog visade R252C‑varianten inte denna vanliga självfosforylering. Istället aktiverade celler som bar EGFR R252C en specifik tillväxtregulator, ERK1/2, mycket starkare än normalt, medan andra klassiska EGFR‑vägar — såsom AKT och STAT3 — i stort sett förblev opåverkade. Att blockera ERK1/2 med en selektiv hämmare utplånade R252C‑cellernas extra tillväxtfördel, vilket bevisar att ERK1/2 är huvudmotorn bakom denna mutants tumördrivande effekt.
Låser receptorn i en ständigt aktiv form
För att förstå hur en yttre förändring kan ge sådan selektiv överaktivitet kombinerade forskarna biokemiska analyser med datorbaserade simuleringar. R252C‑bytet inför en ny cystein på EGFR:s utsida. Två sådana mutanter kan bilda en disulfidbindning — en form av molekylärt häftstift — mellan sina C252‑rester och låsa dem ihop till ett stabilt par. Strukturell modellering visade att denna bindning tvingar receptorernas utsida till en "V‑formad", förskjuten inriktning som starkt efterliknar den aktiva, ligandbundna formen, även utan närvaro av någon tillväxtfaktor. Denna inriktning fortplantas genom membransegmentet och de precis innanförliggande regionerna och vrider de inre enzymdomänerna till en ovanlig konfiguration: de aktiva ytorna pekar in mot cellens inre men hålls för långt isär för att effektivt fosforylera varandra. Istället skapar denna konformation en stark dockningsyta för ERK1/2, vilket gör att EGFR R252C kan fosforylera ERK1/2 direkt och kringgå den vanliga RAS–RAF–MEK‑kaskaden.
Från musmodeller till en enskild patient
Författarna visade att hjärn‑ och lungcancerceller med EGFR R252C växte snabbare i odling och bildade större, mer aggressiva tumörer när de ympades in i möss, jämfört med celler med normalt EGFR. De testade sedan flera generationer av EGFR‑hämmare. Endast afatinib, en andra generations‑hämmare, slog konsekvent ner ERK1/2‑aktivering och minskade markant tumörcelltillväxt. I musemodeller av R252C‑drivna hjärn‑ och lungtumörer bromsade afatinib tumörutbredningen och förlängde överlevnad. Avgörande var att när den ursprungliga patienten — vars sjukdom förvärrats trots cytostatika, ett kärl‑inriktat läkemedel och immunterapi — bytte till afatinib, visade skanningar av både lungor och hjärna tydlig minskning av tumörbördan och patienten fick flera år utan progression.
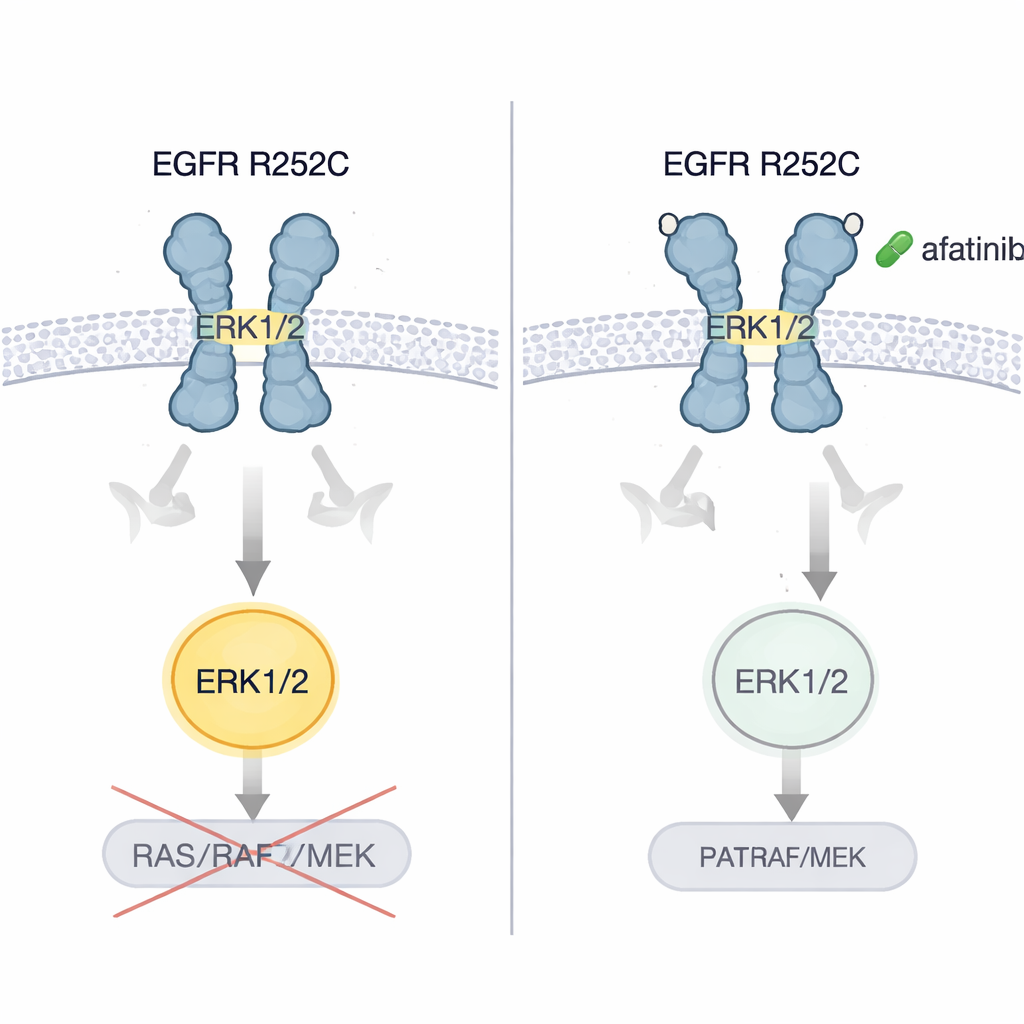
Vad detta betyder för patienter
Detta arbete avslöjar ett tidigare oupptäckt sätt som en cancerframkallande EGFR‑mutation kan verka: genom att häfta ihop två receptorer utanför cellen och vrida dem till en aktiv hållning som direkt slår på ERK1/2 istället för att följa den klassiska signalvägen. För icke‑specialister är huvudpoängen att inte alla mutationer i samma gen beter sig likadant, och att vissa sällsynta förändringar kan svara särskilt väl på specifika befintliga läkemedel. EGFR R252C tycks ge upphov till tumörer som är särskilt känsliga för afatinib. Även om denna slutsats för närvarande vilar på ett detaljerat patientfall plus omfattande laboratoriearbete, pekar den mot mer individualiserad testning av EGFR:s yttre domän‑mutationer och antyder att noga utvalda riktade terapier kan erbjuda nytt hopp för utvalda patienter med svårbehandlade hjärt‑ och lungtumörer.
Citering: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Nyckelord: EGFR‑mutation, gliom, lungcancer, ERK‑signalering, afatinib