Clear Sky Science · sv
Förbättrad polygen prediktion för underrepresenterade grupper genom transferinlärning
Varför ditt DNA-riskpoäng kanske inte fungerar för dig
Genetiska ”riskpoäng” används i allt större utsträckning för att uppskatta en persons sannolikhet att utveckla vanliga tillstånd som diabetes, hjärtsjukdom eller högt blodtryck. Men de flesta av dessa poäng har tagits fram med DNA-data från personer med europeiskt ursprung. Som en följd predicerar de ofta dåligt för personer från andra bakgrunder, vilket väcker frågor om rättvisa och nyttan i verklig klinisk praxis. Denna studie ställer en enkel fråga: kan vi återanvända det vi lärt oss från stora europeiska dataset för att bygga bättre, mer rättvisa genetiska poäng för underrepresenterade grupper—utan att dela någons rådata?
Från miljontals DNA-markörer till en enda riskpoäng
En polygen poäng är som ett betyg som summerar de små effekterna av många genetiska markörer utspridda över genomet. Varje markör får en vikt som återspeglar hur starkt den är associerad med en egenskap, baserat på stora genetiska studier. När dessa studier till övervägande del involverar européer tenderar den resulterande poängen att fungera bäst i just europeiska populationer. Skillnader i genetisk bakgrund—hur vanliga vissa DNA-varianter är och hur de ärvs tillsammans—innebär att samma vikter ofta missar målet i afroamerikanska, hispaniska och andra populationer. Att samla lika stora dataset för varje grupp är dyrt och tidsödande, så författarna vände sig till en maskininlärningsstrategi kallad transferinlärning: i stället för att börja från noll i varje population förfinar de en befintlig modell som tränats någon annanstans.
Hur man lånar kunskap utan att dela rådata
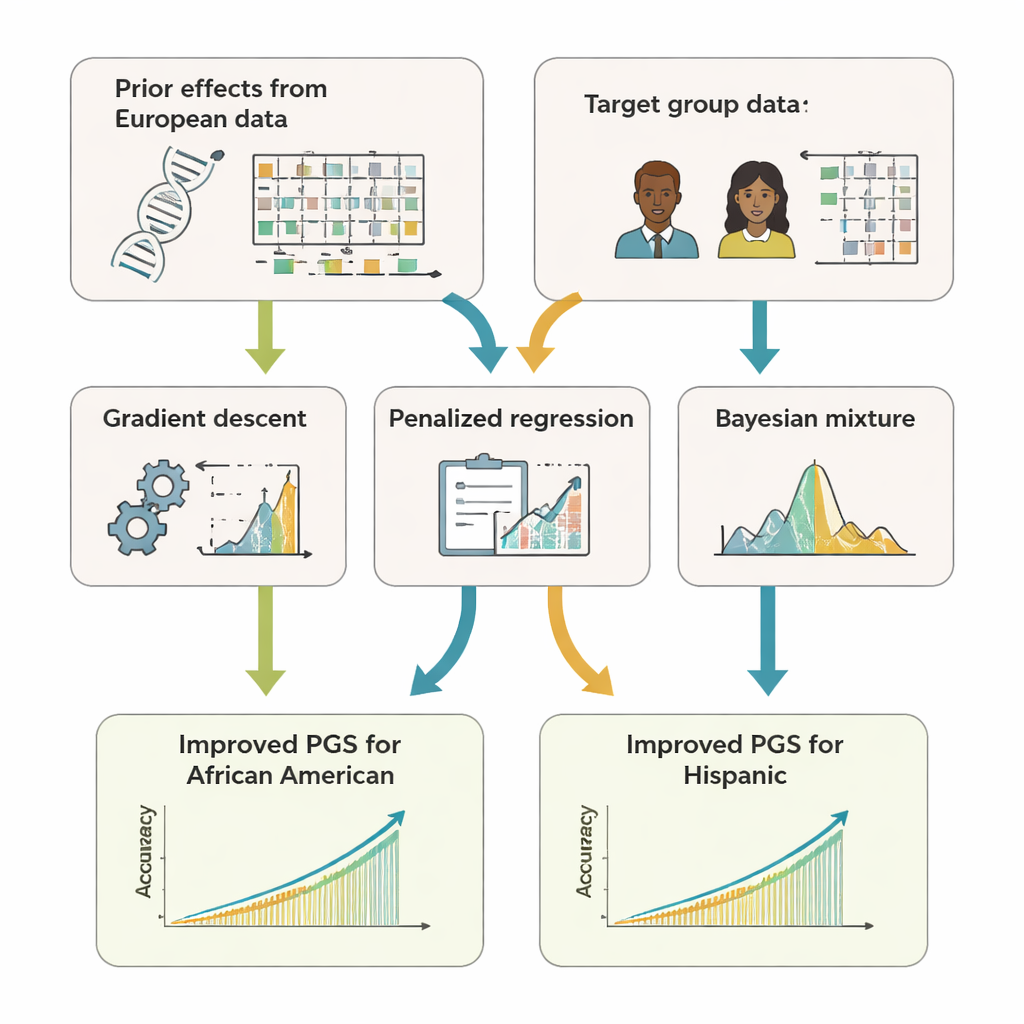
Teamet utvecklade GPTL, ett öppen källkodspaket i R som implementerar tre transferinlärningsmetoder för genetiska poäng. Alla tre utgår från befintliga skattningar av DNA-effekter erhållna i ett stort europeiskt dataset och justerar sedan dessa skattningar försiktigt med hjälp av data från en målgrupp, som afroamerikaner eller hispanics. En metod finjusterar de europeiska vikterna stegvis med gradientnedstigning och stoppar tidigt innan de helt skriver över dem. En andra metod, kallad penaliserad regression, drar aktivt de nya skattningarna mot ursprungsvärdena såvida inte målgruppens data ger starkt stöd för något annat. Den tredje, en bayesisk blandningsmodell, låter varje DNA-markör välja mellan flera informationskällor—såsom flera härstamningsgrupper eller till och med ett ”ingen-effekt”-alternativ—och blandar dem efter hur väl de förklarar målgruppens data.
Sätta metoderna på prov
För att se hur väl dessa angreppssätt fungerar använde författarna både datorbaserade simuleringar och verkliga data från hundratusentals frivilliga i UK Biobank och det amerikanska All of Us-forskningsprogrammet. De fokuserade på afroamerikanska och hispaniska deltagare som målgrupper och använde data från europeisk härkomst som huvudkälla för förhandsinformation. För 11 egenskaper—inklusive längd, kroppsmassindex, blodlipider, blodtryck och njurmarkörer—predicerade transferinlärningspoängen konsekvent bättre än poäng som byggts enbart inom målgruppen eller som återanvänts direkt från européer. Ofta matchade eller överskred deras noggrannhet något den hos mer komplexa ”multi-ancestry”-metoder som kräver att man kombinerar rådata från flera populationer. Avgörande är att GPTL:s metoder bara behöver sammanfattande statistik—aggregerade tal om genetiska effekter—så institutioner kan samarbeta utan att exponera individnivådata.
När mer DNA inte alltid är bättre
Forskarnas analyser tog också upp hur man bäst väljer vilka genetiska markörer som ska ingå. Tvärtemot den vanliga uppfattningen att fler markörer alltid hjälper, fann de att för afroamerikanska och särskilt hispaniska grupper kan inkludering av miljontals mycket svaga signaler faktiskt försämra prestandan, särskilt när man använder starkt förenklade representationer av genetiska korrelationer. Att fokusera på bättre underbyggda markörer och använda rikare information om hur varianter ärvs tillsammans gav ofta mer precisa poäng. Studien visade också att tillskott av förhandsinformation från flera härstamningsgrupper och noggrann modellering av skillnader mellan populationer förbättrade prediktionerna ytterligare.
Vad detta betyder för mer rättvis genetisk riskprediktion
För icke-europeiska populationer kan dagens färdiga genetiska riskpoäng prestera betydligt sämre, vilket potentiellt kan förstärka hälsoojämlikheter. Detta arbete visar att transferinlärning—att smart förfina befintliga Europa-baserade poäng med måttliga dataset från underrepresenterade grupper—kan minska stora delar av den klyftan. I praktiken innebär det att vårdsystem och forskare kan bygga mer precisa och rättvisa genetiska verktyg utan att slå ihop rådata över institutioner eller härkomster, vilket underlättar integritetsfrågor. Ingen enskild metod kommer vara bäst för varje egenskap och population, men GPTL-verktyget visar att mer rättvis genetisk prediktion är tekniskt möjlig om vi behandlar tidigare modeller inte som slutgiltiga produkter utan som utgångspunkter som kan anpassas för alla.
Citering: Wu, H., Pérez-Rodríguez, P., Boehnke, M. et al. Improving polygenic score prediction for underrepresented groups through transfer learning. Nat Commun 17, 1973 (2026). https://doi.org/10.1038/s41467-026-68696-7
Nyckelord: polygena riskpoäng, transferinlärning, genetisk prediktion, hälsoojämlikheter, populationsgenetik