Clear Sky Science · sv
Omfattande jämförelser av metoder för polygena poäng för en- och flervänt härstamning med plattformen PGS-hub
Varför din DNA-riskpoäng spelar roll
Läkarna blir bättre på att läsa vår DNA för att uppskatta vem som löper större risk att utveckla vanliga sjukdomar som hjärt-kärlsjukdom, diabetes eller schizofreni. Dessa uppskattningar, kallade polygena poäng, kombinerar de små effekterna från många genetiska varianter till ett enda tal. Men det finns numera många konkurrerande sätt att beräkna sådana poäng, och de fungerar inte lika bra för personer med olika härstamningsbakgrund. Denna studie syftade till att jämföra ledande metoder direkt mot varandra och att bygga en onlinetjänst, PGS-hub, som låter forskare beräkna dessa poäng på ett konsekvent och enkelt sätt.
En enda plats för DNA-riskberäknare
Författarna skapade PGS-hub, en webbplattform som döljer mycket av den tekniska komplexiteten bakom polygena poäng. Användare laddar upp genetiska studiedata som sammanfattar hur miljontals DNA-markörer relaterar till en sjukdom eller egenskap. De väljer sedan den härstamningsbakgrund för populationen de är intresserade av—till exempel europeisk eller afrikansk—och väljer från en meny av populära beräkningsmetoder. I bakgrunden konverterar PGS-hub indata till rätt format, kopplar in förbyggda referenspaneel som beskriver hur närliggande DNA-markörer korrelerar, och kör ett stort antal jobb på ett högpresterande beräkningssystem. Resultatet är en kompakt fil med vikter som kan appliceras på individuella genom för att generera en poäng för varje person. 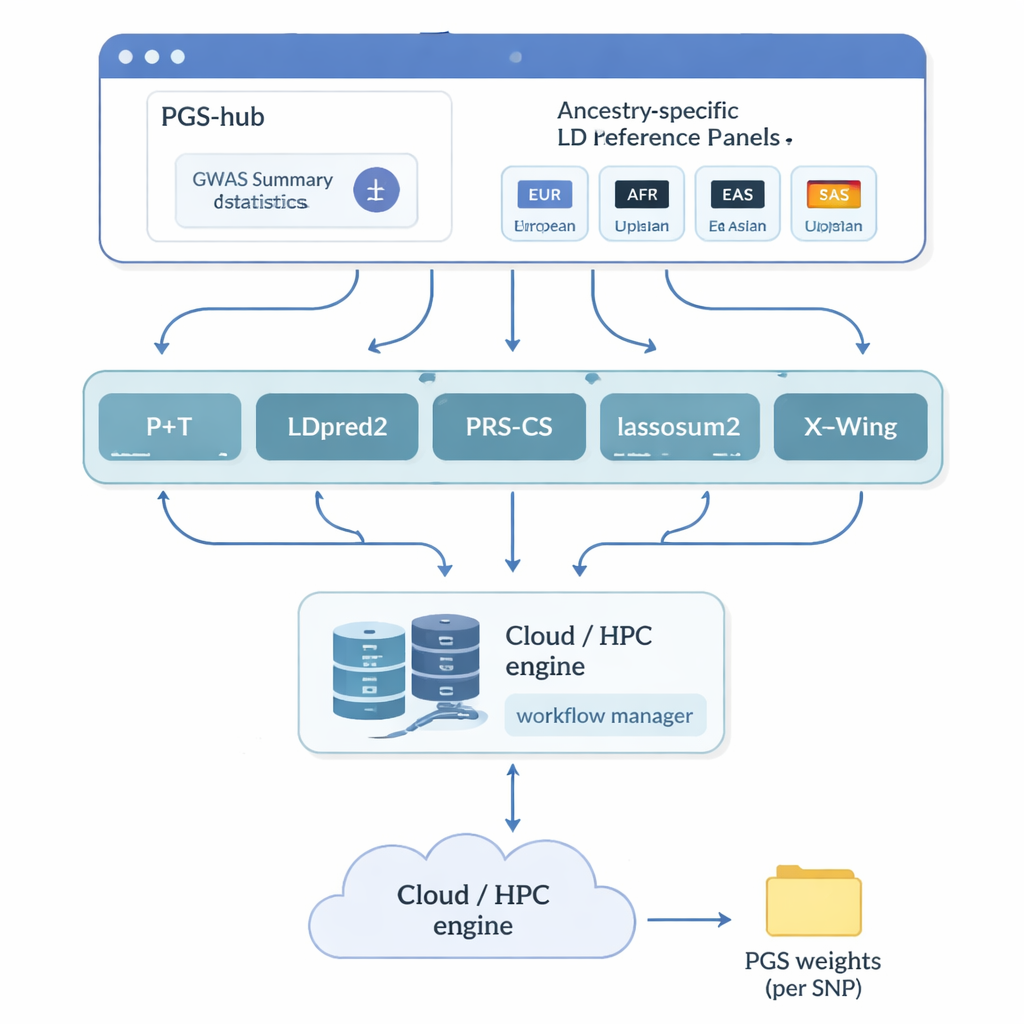
Att testa 13 beräkningsmetoder
För att se vilka angreppssätt som fungerar bäst jämförde gruppen 13 toppmoderna metoder över 36 sjukdomar och egenskaper i nästan 380 000 personer med europeisk härstamning och knappt 8 000 personer med afrikansk härstamning från UK Biobank. De utvärderade inte bara hur väl varje poäng predicerade vem som hade en sjukdom eller högre värde på en egenskap, utan också hur mycket beräkningstid och minne varje metod krävde. Bland européerna levererade en metod kallad LDpred2 i allmänhet de mest exakta poängen och slog ofta andra med en tydlig marginal. Ett fåtal alternativ—lassosum2, PRS-CS och SDPR—presterade nästan lika bra för många egenskaper, medan några äldre metoder halkade efter. För egenskaper som längd eller Crohns sjukdom förklarade de bästa poängen en avsevärd del av den genetiska risken; för andra, som njurfunktion, hade alla metoder svårt att prestera väl, vilket speglar svagare underliggande genetiska signaler.
Insikter för olika populationer och kombinerade metoder
En stor oro inom genetisk prediktion är att metoder som framför allt tränats i europeiska populationer kanske inte överförs väl till människor med andra härstamningar. När författarna upprepade sina jämförelser med genetiska studier från personer med afrikansk härstamning presterade alla metoder sämre, vilket framhäver bristen på stora studier i dessa populationer. Ändå tenderade LDpred2 och SDPR att vara bland de bättre alternativen. Gruppen undersökte också ”multi-härstamnings”-metoder som uttryckligen kombinerar information över populationer. Här överträffade en relativt enkel strategi—linjärt kombinera de bästa härstamningsspecifika LDpred2-poängen till en enda LDpred2-multi-poäng—mer komplicerade multi-härstamhetsmodeller som PRS-CSx och X-Wing för både europeiska och afrikanska grupper. Dessutom visade författarna att bygga ett ensemble, som blandar de starkaste poängen från flera metoder, ytterligare förbättrade prediktionen över alla egenskaper, särskilt för högärftliga sjukdomar som schizofreni och kranskärlssjukdom. 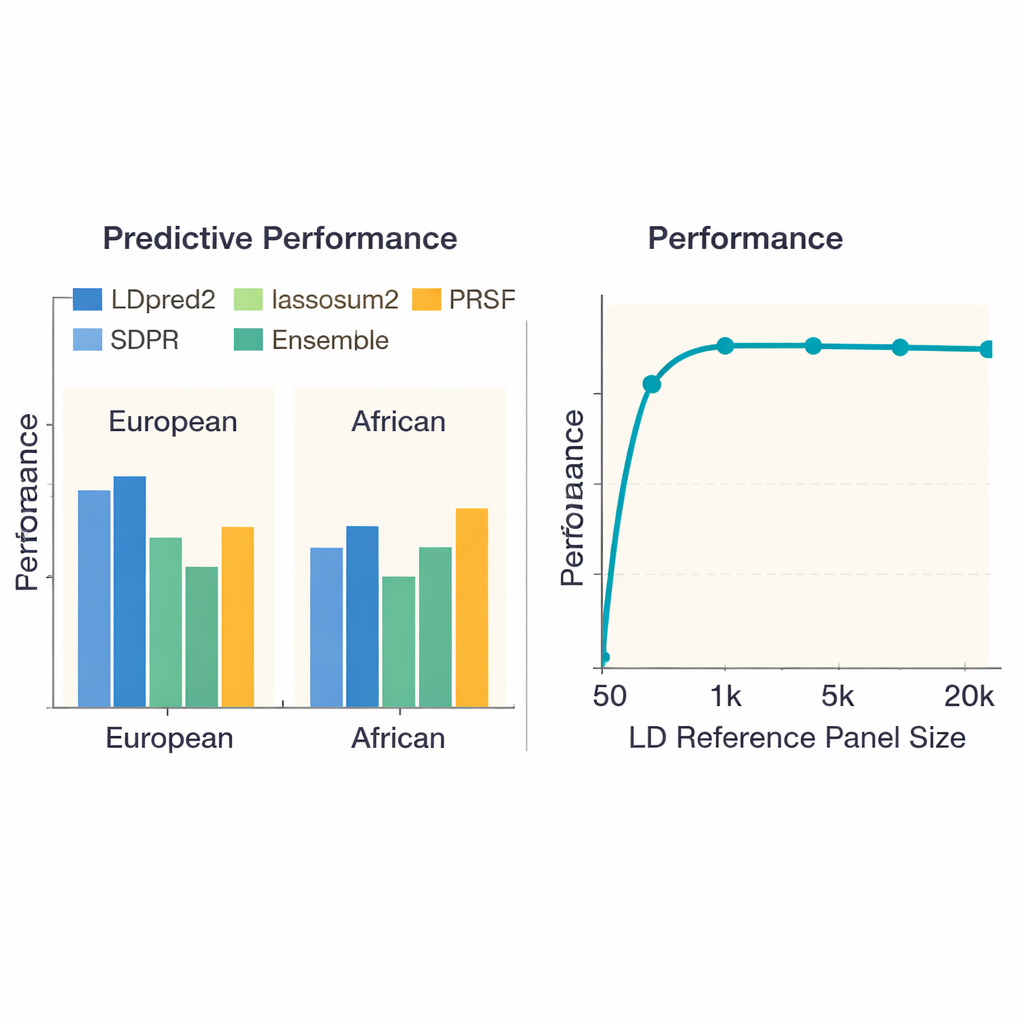
Hur dataval och beräkningsgränser påverkar poängen
Studien undersökte hur storleken på referenspanelet—mängden personer som används för att lära hur närliggande DNA-markörer samvarierar—påverkade prestanda. När detta panel var mycket litet (färre än 1 000 individer) var poängen märkbart mindre precisa. När panelen växte till omkring 5 000 personer förbättrades prestandan kraftigt för att sedan plana ut, vilket antyder att ständigt större paneler ger avtagande avkastning. Förvånande nog hjälpte det inte alltid att bara lägga till fler DNA-markörer: att använda omkring 6,6 miljoner varianter försämrade ibland prognoserna jämfört med att använda ett noggrant utvalt set på ungefär 1,1 miljoner, sannolikt eftersom extra markörer tillförde mer brus än nyttig signal. Författarna dokumenterade också stora skillnader i beräkningskostnad. Enkla metoder som grundläggande pruning-and-thresholding blev klara på under en timme per egenskap, medan vissa bayesiska angreppssätt krävde hundratals CPU-timmar—information som är viktig för stora projekt eller grupper med begränsade resurser.
Vad detta betyder för framtida DNA-baserad prediktion
För icke-specialister är huvudbudskapet att inte alla DNA-riskpoäng är likadana, och detaljerna i hur de byggs påverkar starkt vem som gynnas av dem. Detta arbete ger praktisk vägledning: metoder som LDpred2 och välkonstruerade ensembler tenderar att ge de mest pålitliga prediktionerna i stora europeiska dataset, och multi-härstamningskombinationer kan överträffa mer komplexa modeller över populationer. Samtidigt understryker den minskade noggrannheten för personer med afrikansk härstamning ett akut behov av större och mer varierade genetiska studier. Genom att paketera många metoder i en enda, standardiserad onlineplattform sänker PGS-hub tröskeln för forskare världen över att generera och jämföra polygena poäng—ett viktigt steg mot att använda sådana poäng rättvist och effektivt inom medicinen.
Citering: Chen, X., Wang, F., Zhao, H. et al. Comprehensive benchmarking single and multi ancestry polygenic score methods with the PGS-hub platform. Nat Commun 17, 2014 (2026). https://doi.org/10.1038/s41467-026-68599-7
Nyckelord: polygena poäng, genetisk riskprediktion, PGS-hub-plattformen, genomik för flera härstamningar, UK Biobank