Clear Sky Science · sv
TET1 som en huvudregulator som kontrollerar GPX4-beroende och -oberoende ferroptosövervakning i akut myeloisk leukemi
Varför denna forskning är viktig för cancerbehandling
Många nya cancerläkemedel syftar till att tvinga maligna celler in i ett självförstörande läge kallat ferroptos, en form av celldöd driven av järn och lipidskador. Ändå motstår vissa tumörer denna strategi. Denna studie visar hur ett DNA‑modifierande protein kallat TET1 hjälper leukemiceller att undvika ferroptos genom två separata biokemiska försvarssystem — och visar att blockering av dessa försvar kan göra även resistenta tumörer sårbara.
En dödlig blandning av järn och skadade fetter
Ferroptos uppstår när järn driver okontrollerad oxidation av fetter i cellmembran, vilket slutligen får cellerna att brista. I akut myeloisk leukemi (AML), liksom i många andra cancerformer, använder cellerna kraftfulla övervakningssystem för att hålla denna process i schack. En nyckelvakt är enzymet GPX4, som använder ett litet molekylärt ämne kallat glutation för att neutralisera skadliga lipidperoxider. Andra reservsystem genererar antioxidantmolekyler som kan fånga farliga radikaler även när GPX4 är komprometterat. Att förstå vilka huvudbrytare som koordinerar dessa försvar är avgörande för att utforma terapier som pålitligt utlöser ferroptos i cancerceller samtidigt som friska vävnader skonas.
TET1 framträder som en central kontrollpunkt
Forskarna jämförde dussintals cancercellprover, inklusive många AML‑linjer och patient‑ härledda celler, och noterade ett tydligt mönster: celler som motstod ferroptos hade högre nivåer av TET1, ett enzym som ändrar DNA:s kemiska märken och påverkar genaktivitet. När de minskade TET1‑nivåerna med genetiska verktyg eller hämmade dess aktivitet med en liten molekyl blev cancercellerna markant mer känsliga för ferroptos‑utlösande läkemedel. Detta gällde både i odlingskärl och i musmodeller av AML. Omvänt skyddade ökad TET1‑uttryck cellerna från ferroptotisk död och begränsade uppbyggnaden av reaktiva syrearter, de kemiskt aggressiva biprodukter som driver membranskada.
Förstärkning av huvudantioxidantskölden
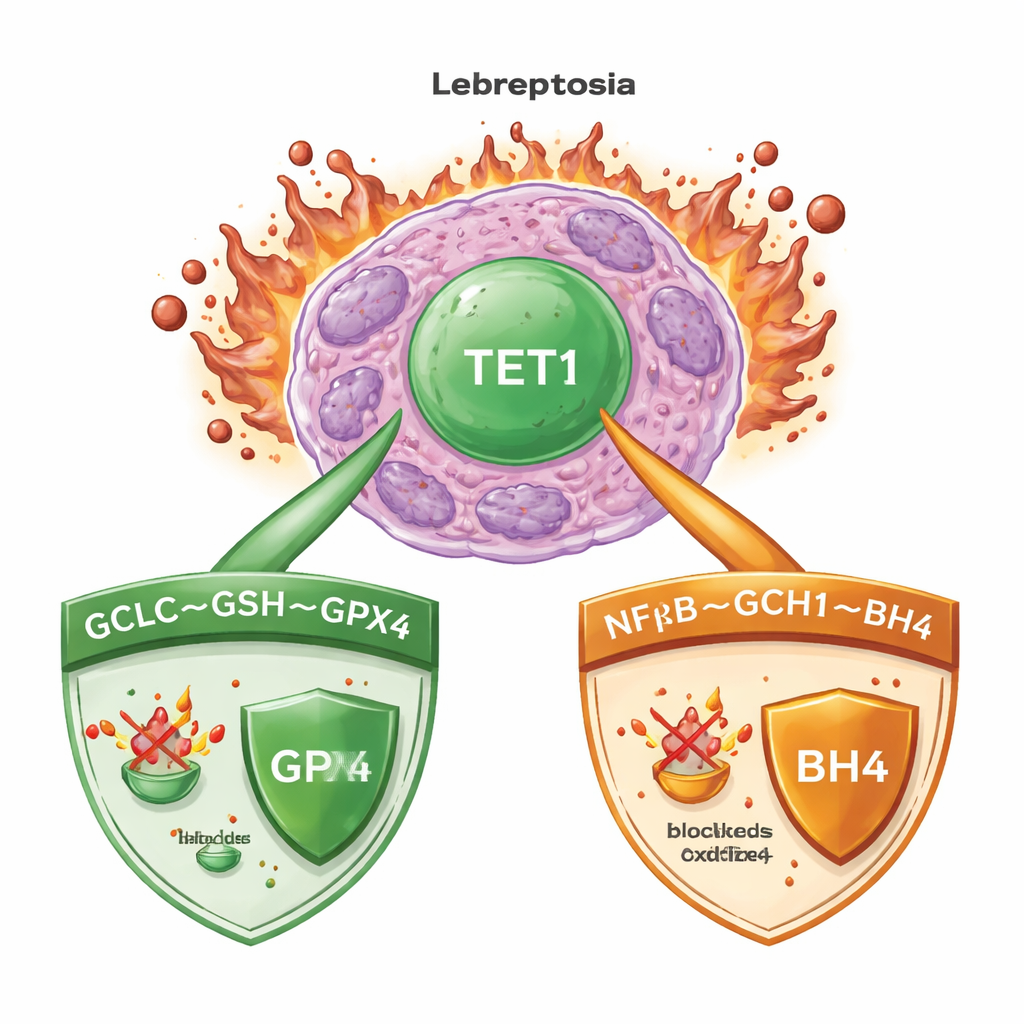
När teamet gick djupare kartlade de var TET1 verkar på genomet och fann att det direkt aktiverar en gen kallad GCLC. GCLC kodar för ett kritiskt enzym som startar produktionen av glutation, bränslet för GPX4. Genom att öka en specifik DNA‑markör (5‑hydroxymetylcytosin) vid GCLC‑promotorn spär TET1 på glutationsyntesen. Under normala näringsförhållanden ökar detta cellens huvudsakliga antioxidantpool. Vid cystinbrist tillverkar samma enzymkomplex också ovanliga γ‑glutamyl‑peptider som hjälper till att ta upp överskott av glutamat, ett annat sätt att dämpa ferroptos. Både i odlade celler och i möss ledde förlust av TET1 eller farmakologisk hämning av glutationsyntesen till kraftigt sänkta nivåer av glutation och dessa skyddande peptider, vilket gjorde leukemiceller mycket mer sårbara för ferroptos‑utlösare.
En andra, GPX4‑oberoende flyktrutt
Överraskande nog slutade inte TET1:s skyddande roll vid glutation–GPX4‑axeln. Även när GPX4 självt togs bort från leukemiceller kunde extra TET1 fortfarande förhindra ferroptotisk död, vilket antyder en andra försvarslinje. Författarna spårade detta tillbaka till TET1:s aktivering av NFκB‑signalvägen, i synnerhet en komponent kallad NFKB2. Detta ökar i sin tur uttrycket av GCH1, ett enzym som producerar antioxidantmolekylen BH4. BH4 kan skydda membranlipider från oxidation utan att förlita sig på GPX4. När GCH1 tystades genetiskt eller kemiskt blockerades förlorade TET1 delvis sin förmåga att skärma av cellerna från ferroptos. Tillsammans definierar dessa fynd en TET1–NFKB2–GCH1‑väg som utgör ett GPX4‑oberoende system för ferroptosövervakning.
Att göra en svaghet till en terapeutisk möjlighet
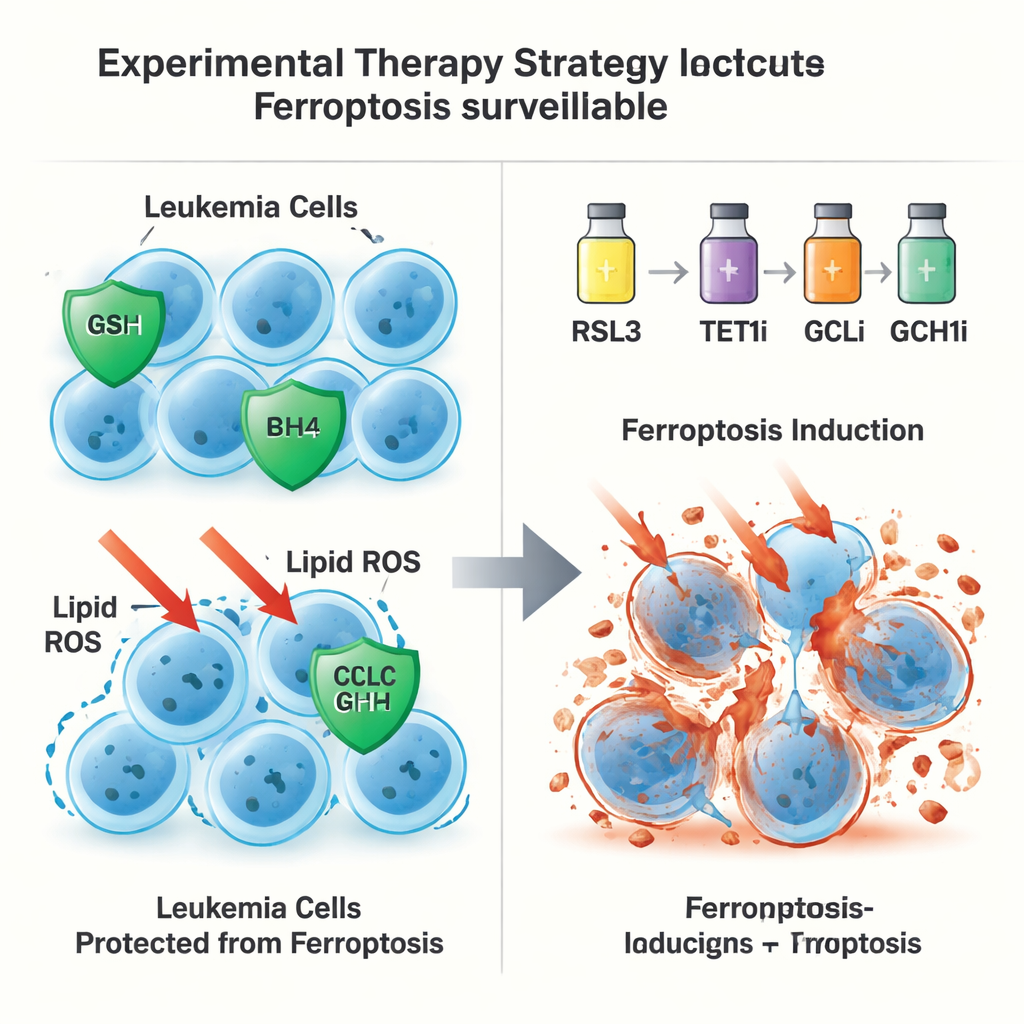
Med denna dubbelriktade karta testade forskarna om samtidig stimulering av ferroptos och försvagning av TET1‑kontrollerade försvar kunde ge en terapeutisk fördel. I musmodeller för AML och patient‑härledda leukemitransplantat i möss reducerade låga doser av ett ferroptos‑utlösande läkemedel kombinerat med hämmare av TET1, GSH‑syntes (via GCLC) eller GCH1 kraftigt leukemibördan, förlängde överlevnad och utarmade populationer av leukemistartande celler. Viktigt är att ferroptosutlösaren användes i en bråkdel av de doser som rapporterats i tidigare studier, vilket minskar oro för toxicitet mot normala blodstamceller.
Vad detta betyder för framtida cancerbehandlingar
För icke‑specialister är huvudbudskapet att leukemiceller överlever genom att driva två överlappande antioxidant"sköld"system, båda koordinerade av TET1: ett centrerat på glutation och GPX4, och ett annat baserat på GCH1 och BH4. Denna studie visar att genom att måttligt aktivera ferroptos samtidigt som man blockerar TET1 och dess nedströms‑partner kan kliniska behandlingar en dag övervinna resistens och selektivt pressa cancerceller över gränsen, utan att överväldiga friska vävnader. Även om dessa strategier ännu inte är redo för klinisk användning identifierar arbetet TET1 som en kraftfull kontrollnod och ett lovande mål för kombinationsbehandlingar vid AML och potentiellt andra svårbehandlade cancerformer.
Citering: Yang, L., Lu, J., Yun, W. et al. TET1 as a master regulator controlling GPX4-dependent and -independent ferroptosis surveillance in acute myeloid leukemia. Nat Commun 17, 1800 (2026). https://doi.org/10.1038/s41467-026-68509-x
Nyckelord: ferroptos, akut myeloisk leukemi, TET1, glutation, cancerepigenetik