Målstyrda läkemedel har förändrat behandlingen av vissa lungcancerformer genom att rikta in sig på en felaktig tillväxtsignal kallad EGFR. För de flesta patienter slutar dessa läkemedel dock fungera inom ett par år eftersom tumören utvecklar resistens. Denna studie avslöjar en överraskande vändning: när tumörer blir resistenta mot EGFR‑hämmare utvecklar de en ny akilleshäl som kan attackeras med en annan typ av förening. Att förstå denna dolda svaghet kan ge uppslag till framtida behandlingsstrategier som tränger in i cancerns evolution i stället för att hela tiden jaga den.
En dold svaghet avslöjad
Forskarnas fokus låg på icke‑småcellig lungcancer som drivs av mutant EGFR, en vanlig form av sjukdomen. I laboratoriet jämförde de läkemedelskänsliga cancerceller med närbesläktade celler som utvecklat resistens mot EGFR‑blockerande läkemedel som gefitinib och osimertinib. De testade sedan ett bibliotek med cirka 2 100 små molekyler för att se vilka som dödade de resistenta cellerna mer effektivt än de ursprungliga, läkemedelskänsliga cellerna. Bland många kandidater utmärkte sig en förening, kallad MCB‑613, konsekvent. Resistent‑celler som avfärdade EGFR‑hämmare visade sig vara ovanligt sårbara för MCB‑613, både i petriskålar och i mus‑tumörer.
Fånga blandade tumörpopulationer Figure 1.
Riktiga tumörer är blandningar av celler: vissa förblir känsliga för det ursprungliga läkemedlet medan andra förvärvar resistens genom olika genetiska knep. Teamet undersökte om en kombination av en EGFR‑hämmare och MCB‑613 kunde utplåna denna mångfald. I ett kontrollerat experiment blandade de till största delen läkemedelskänsliga celler med en liten andel av flera resistenta typer, för att efterlikna en patients tumör. Behandling av denna blandade population med antingen EGFR‑hämmaren eller MCB‑613 ensam tillät några celler att överleva och växa. Men när båda medlen användes tillsammans kollapsade hela populationen. Detta tyder på att man genom att para ett standardiserat målinriktat läkemedel med ett noggrant utvalt ”kollateral känslighets”‑läkemedel kan driva tumörer in i ett evolutionärt dödsspår.
En molekylär bro som bryter en väktare
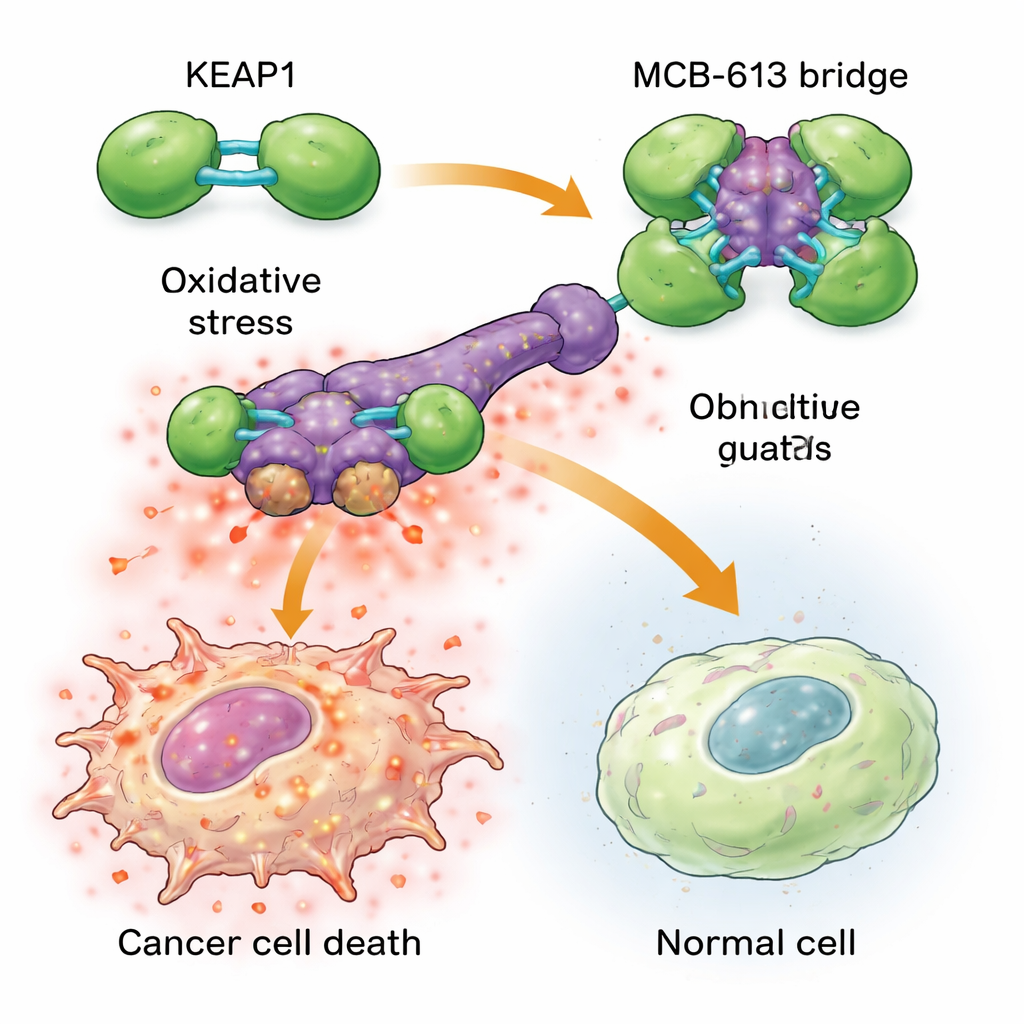
För att förstå varför MCB‑613 slår så hårt mot resistenta celler undersökte forskarna vilka proteiner det binder till. Genom kemiska prober och ett riktat CRISPR‑skärningsscreen identifierade de ett protein kallat KEAP1 som avgörande för MCB‑613:s effekt. KEAP1 fungerar normalt som en cellulär väktare, som känner av stress och hjälper till att reglera skyddande svar. Teamet fann att MCB‑613 fäster vid KEAP1 på ett ovanligt sätt: det beter sig som en stel molekylär bro som länkar KEAP1‑enheter tillsammans till överdimensionerade, abnorma kluster. Denna process beror inte på de vanliga reaktiva svavelinnehållande platserna i KEAP1, utan på en specifik lysin‑aminosyra i dess dimeriseringsregion. När den lysinen muterades kunde MCB‑613 inte längre klumpa ihop KEAP1, och de resistenta cellerna var inte längre hypersensitiva för föreningen.
Att vända hjälpsam stress till dödlig överbelastning Figure 2.
KEAP1‑klumpning sätter igång en farlig kedjereaktion inne i läkemedelsresistenta cancerceller. Dessa celler lever redan under högre bakgrundsstress, med förhöjda nivåer av reaktiva syrearter (skadliga kemiska biprodukter) och ökad aktivitet i ett skyddande signalsystem känt som det integrerade stresssvaret. När MCB‑613 tillsätts pressar störningen av KEAP1 denna stressade status över gränsen: reaktiva syrearter byggs upp ytterligare och viktiga stressregulatorer kallade ATF4 och CHOP slår på kraftfulla dödsprogram. Att blockera dessa stressregulatorer, eller kemiskt avlägsna reaktiva syrearter, skyddade till stor del cellerna från MCB‑613. Intressant nog var den klassiska KEAP1‑partnern NRF2, ofta betraktad som huvuddrivaren för antioxidantförsvar, inte ansvarig för dödandet; att ta bort NRF2 gjorde till och med cellerna ännu mer känsliga, vilket understryker att MCB‑613 utnyttjar en annan, icke‑kanonisk väg.
Vad detta kan innebära för framtida behandlingar
För närvarande är MCB‑613 i sig ett verktygssubstans med kemiska nackdelar som gör den olämplig som läkemedel. Men den avslöjar ett kraftfullt koncept: när lungcancer utvecklar resistens mot EGFR‑hämmare kan den bli fastlåst i ett stressat tillstånd som selektivt kan riktas mot med föreningar som tvingar KEAP1 in i dysfunktionella sammankopplingar. I princip skulle förfinade versioner av sådana ”molekylära broar” kunna utvecklas för att bli säkrare och mer precisa, vilket ger onkologer ett sätt att styra tumörer till ett ”omöjligt val” mellan känslighet för den ursprungliga målinriktade terapin och känslighet för den uppföljande stressframkallande agenten. Denna evolutionära inhägningsstrategi skulle så småningom kunna hjälpa till att fördröja eller övervinna resistens vid EGFR‑mutant lungcancer och möjligen även andra svårbehandlade cancerformer.
Citering: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1