Clear Sky Science · sv
DOT1L-aktivitet begränsar transkriptionens förlängningshastighet och gynnar RNAPII-pausning för att underlätta mutagenes av AID
Hur våra immunceller finjusterar riskfyllda DNA-ändringar
Vårt immunsystem framställer potenta antikroppar genom att medvetet mutera sitt eget DNA, en riskfylld strategi som ibland kan främja cancer. Denna studie ställer en förledande enkel fråga med stora följder: vad bestämmer var, och hur effektivt, dessa avsiktliga mutationer uppstår? Svaret kretsar kring ett protein kallat DOT1L, som justerar hastigheten för genläsning i B-celler och därigenom hjälper till att rikta mutationsmaskineriet mot rätt platser.
Att slå på mutation för att förbättra antikroppar
När B-celler möter en infektion uppgraderar de sina antikroppar på två sätt. De inför små förändringar i antikroppens bindningsregion för att förbättra hur hårt den binder patogener, och de byter ut antikroppens svans för att ändra hur immunsystemet svarar. Båda uppgraderingarna startar med ett enzym kallat AID, som nickar och ändrar DNA i aktivt transkriberade gener. Medan AID är avgörande för ett gott immunsvar kan det också påverka andra gener och skapa farliga brott som driver blodcancer. Tidigare arbete visade att AID föredrar gener som transkriberas mycket och regleras av kraftfulla DNA-switchar kallade super‑enhancers, men detta förklarade inte fullt ut varför endast ett utvalt delmängd av gener verkligen är sårbara.
En kromatinmarkör som flaggar AID-känsliga gener
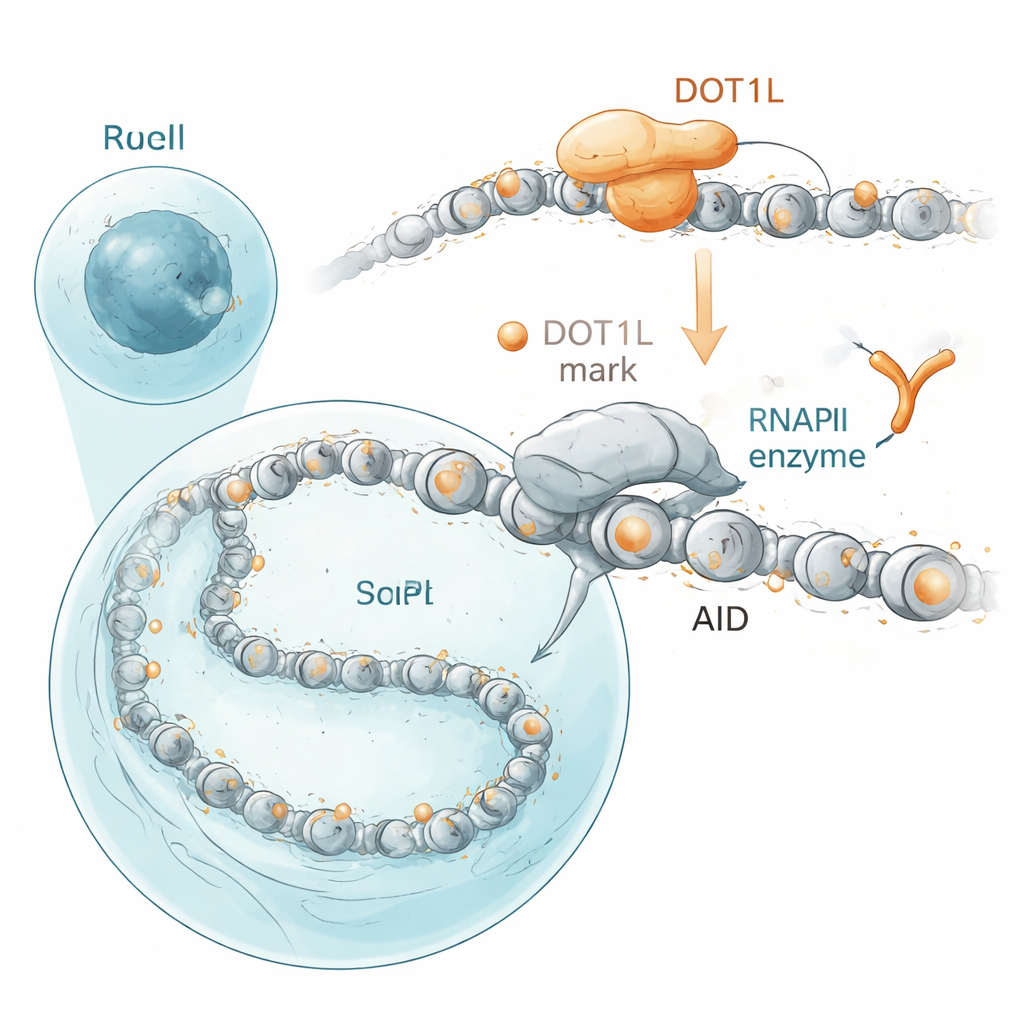
Forskarnas fokus riktades mot proteiner som sitter nära AID i nukleus. Med en proximetetsmärkningsteknik i humana celler fann de att AID klustrar nära DOT1L, ett enzym som sätter en specifik kemisk mark på histonproteinerna kring vilka DNA är lindat. Denna märkning, en kemisk etikett på histon H3 vid position K79, är vanlig på aktiva gener. I mus‑B‑celler bär gener som AID ofta muterar—inklusive antikroppsgener och cancerassocierade off‑targets—särskilt höga nivåer av dessa DOT1L‑skapade markeringar. När forskarna inaktiverade DOT1L i B‑cellinjer eller blockerade dess aktivitet med ett läkemedel minskade antikropparnas "class switching", liksom AID‑drivna DNA‑brott och cancerbenägna fusioner mellan antikroppsgener och tillväxtgenen cMyc. Viktigt är att DOT1L:s katalytiska funktion, inte bara dess närvaro, krävdes: mutanta varianter som inte längre kunde sätta histonmarken lyckades inte återställa normal antikropps‑switching.
Att bromsa genläsaren för att ge AID tid
Vid första anblicken är detta förvånande eftersom DOT1L förknippas med aktiva gener, ändå ledde dess förlust inte helt enkelt till att gener stängdes av. Med en metod som fångar nyligen skapad RNA upptäckte teamet att B‑celler utan DOT1L faktiskt producerade mer nascent transkript över många DOT1L‑markerade gener—even though there was slightly less of the main gene-reading enzyme, RNA polymerase II, sitting on those genes. Genom att kombinera denna nascenta RNA‑avläsning med kartor över polymeras‑okupans drog de slutsatsen att under normala förhållanden agerar DOT1L:s histonmarkeringar som milda hastighetsgupp. De saktar ner polymeraset när det rör sig längs genen och förlänger korta pauser nära startpunkten och inom kroppen av DOT1L‑markerade gener. Utan DOT1L springer polymeraset snabbare och pauserna blir kortare. Eftersom AID behöver korta fönster där DNA är exponerat och polymeraset dröjer vidtar paradoxalt nog denna acceleration AID:s förmåga att fästa och utföra sitt jobb, även när den totala transkriptionen ökar.
Att koppla isär genaktivitet från mutationsrisk
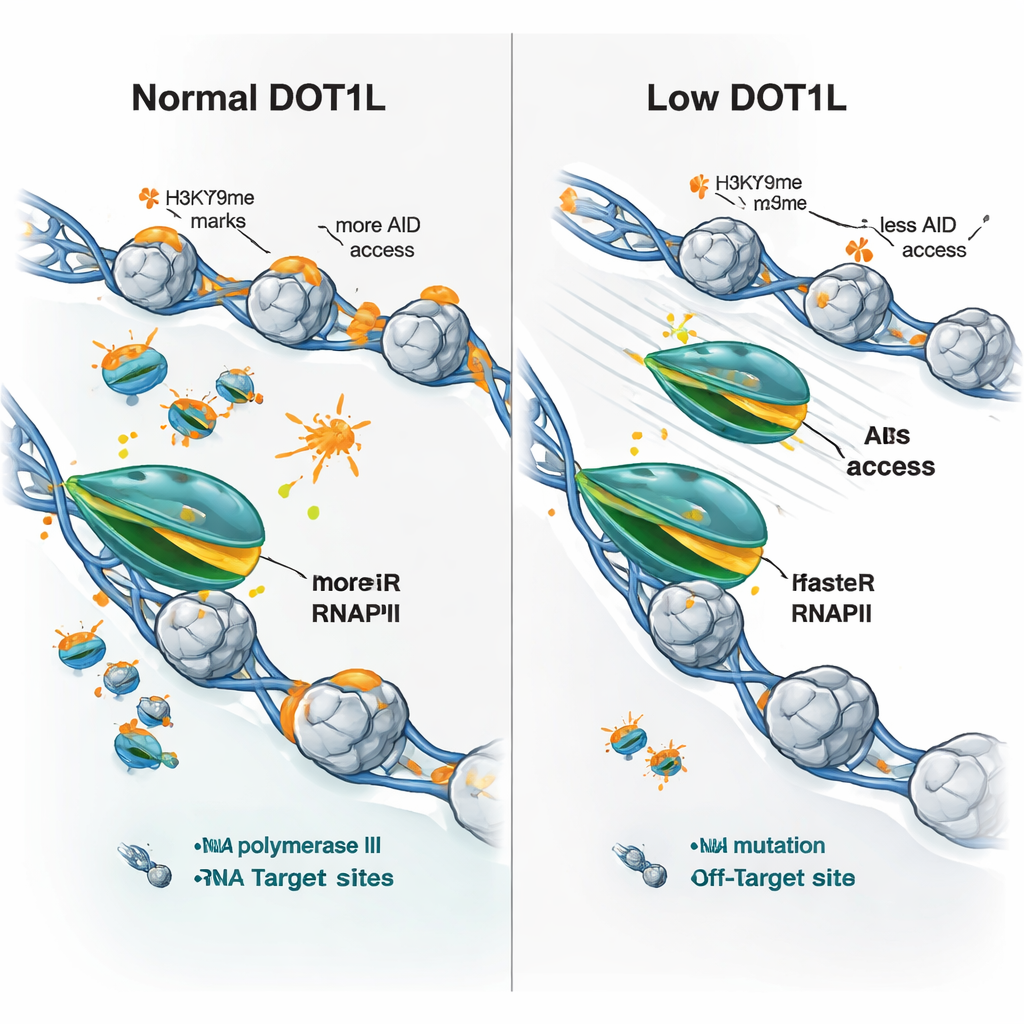
Forskarna frågade sedan om dessa hastighetsförändringar kunde förklara det blandade mönstret av genuttryck som ses när DOT1L saknas—vissa gener ökar, andra minskar. De fann att praktiskt taget alla DOT1L‑markerade gener delade en egenskap i knockout‑celler: snabbare förlängning av RNA‑polymeras II. Men utfallet berodde på utgångstillståndet. Långsamma, svagt uttryckta gener tenderade att producera mer RNA när polymeraset snabbade upp, medan mycket aktiva, långa gener med redan snabba polymeraser ibland producerade mindre, sannolikt eftersom för snabb passage stör effektiv bearbetning eller slutförande. Avgörande var att vid både antikroppsgener och klassiska AID‑off‑targets ledde DOT1L‑förlust till snabbare polymerasrörelse, mindre bevis för polymerasets "stopp", och signifikant minskad AID‑okupans, även när generna i sig inte nedreglerades.
Varför detta är viktigt för immunitet och cancer
Tillsammans målar arbetet upp DOT1L som en subtil trafikregulator för maskineriet som läser av gener i B‑celler. Genom att installera specifika histonmarkeringar bromsar DOT1L svagt RNA‑polymeras II och förlänger dess pauser, vilket skapar en transkriptionell miljö där AID kan engageras produktivt i antikroppsgener—och tyvärr i en begränsad uppsättning andra sårbara gener—för att införa mutationer. När DOT1L saknas eller hämmas rusar polymeraset igenom och lämnar AID färre chanser att verka, vilket dämpar antikroppsdiversifiering och samtidigt minskar risken för vissa skadliga omarrangemang. Denna mekanistiska insikt förklarar varför förlust av DOT1L både kan öka och minska genuttryck, och kopplar den fina kontrollen av transkriptionshastighet direkt till var vårt immunsystem vågar skriva om sitt eget DNA.
Citering: Subramani, P.G., Seija, N., Ridani, J. et al. DOT1L activity limits transcription elongation velocity and favors RNAPII pausing to facilitate mutagenesis by AID. Nat Commun 17, 1623 (2026). https://doi.org/10.1038/s41467-026-68332-4
Nyckelord: antikroppsdiversifiering, AID-enzymet, DOT1L, geners transkription, B-celler