Clear Sky Science · sv
Glycerol-3-fosfat-acyltransferas förvärrar α-synuklein-inducerad toxicitet genom att öka lipidperoxidation
Varför fett i hjärnan spelar roll för Parkinsons
Parkinsons sjukdom beskrivs vanligtvis som ett problem med ett protein kallat alpha‑synuklein som klumpar sig och skadar hjärnceller som styr rörelse. Denna studie visar att hjärnans fetter — särskilt hur de bildas och skadas — spelar en överraskande viktig roll för hur giftigt alpha‑synuklein blir. Genom att identifiera ett fettbildande enzym som förvärrar nervcellsskada pekar arbetet ut en ny, läkemedelsbar väg som kan komplettera befintliga försök att angripa Parkinsons vid dess rötter.
Ett protein som beter sig illa vid Parkinsons
Människor med Parkinsons förlorar successivt dopaminproducerande nervceller i en djup hjärnregion som koordinerar rörelse. Inuti dessa döende celler hittar forskare ofta täta avlagringar kallade Lewy-kroppar, fyllda med proteinet alpha‑synuklein. I sällsynta familjer orsakar mutationer eller extra kopior av alpha‑synuklein-genen direkt Parkinsons, men vanliga genetiska varianter i den genen ökar bara risken måttligt. Det tyder på att andra gener och vägar modifierar hur skadligt alpha‑synuklein blir. I allt större utsträckning pekar bevis mot lipider — fetter och fettliknande molekyler som bygger cellmembran och energilager — som viktiga medspelare i både alpha‑synuklein‑klumpning och nervcellernas död.
Att hitta ett kraftfullt lipidenzym i bananflodsmodeller
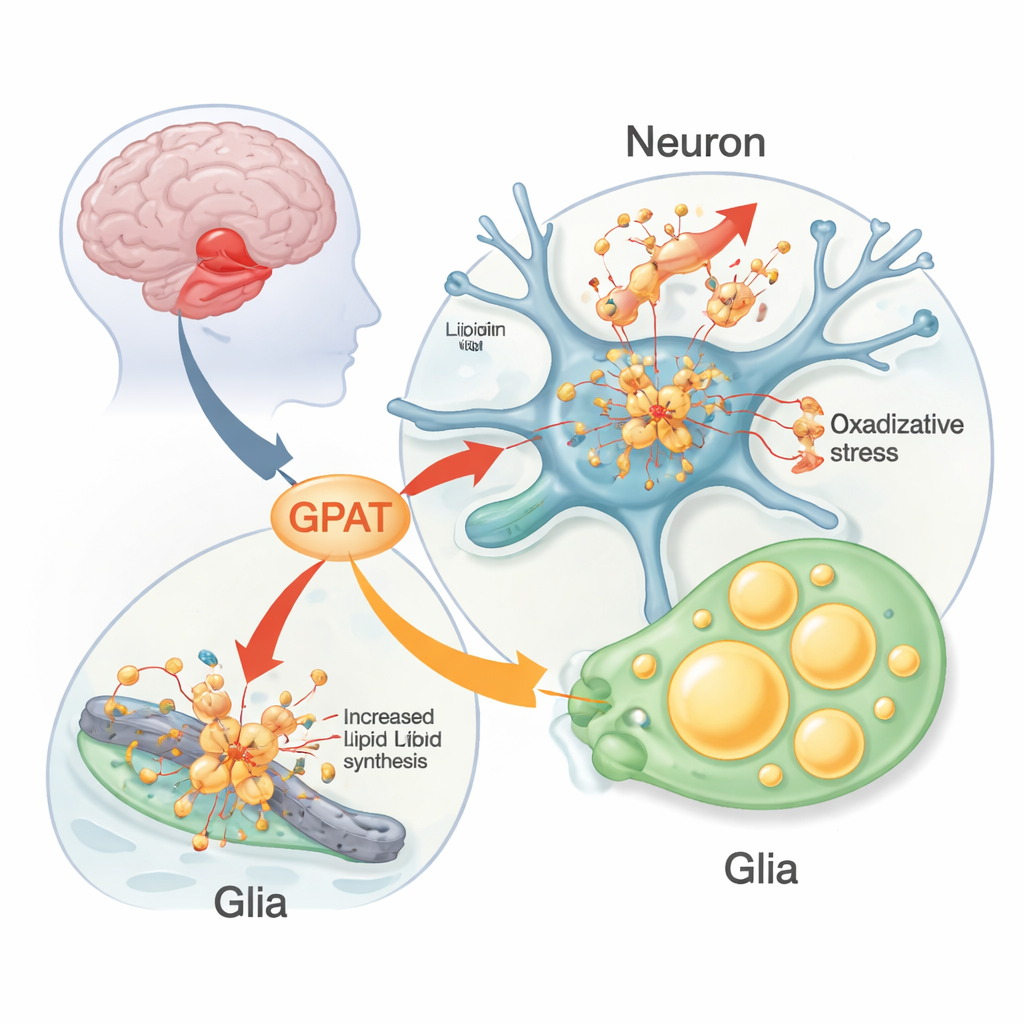
Forskarna använde bananflugor konstruerade för att producera mänskligt alpha‑synuklein i sitt nervsystem som en levande försöksmodell. Dessa flugor utvecklar Parkinson‑lika problem: förlust av dopaminproducerande neuroner, svårigheter att klättra och störda dagliga aktivitetsrytmer. Teamet ändrade systematiskt flugans varianter av mänskliga Parkinson‑riskgener för att se vilka som påverkade alpha‑synukleins verkningar. En kandidat stack ut: en gen kallad mino, som kodar för en mitokondriell form av enzymet glycerol-3-fosfat-acyltransferas (GPAT). GPAT sitter vid ingångspunkten för byggandet av fosfolipider och triglycerider — de lipider som bildar membran och fettdroppar. När teamet minskade mino-aktiviteten i neuroner bevarade alpha‑synuklein‑flugorna fler av sina dopaminneuroner och rörde sig bättre längre; att öka mino gav motsatt, skadlig effekt.
Skadade fetter, stressade mitokondrier och gliala fettdroppar
När de gick djupare fann forskarna att GPAT påverkade hur mycket oxidativ skada som byggdes upp i hjärnans lipider. I alpha‑synuklein‑flugor som hölls vid högre temperatur (vilket förvärrar sjukdomsdrag) ökade lipidperoxidation — fettens kemiska ”rostning” — i hjärnmembran. Att sänka mino minskade denna skada, medan överexpression ökade den; utan alpha‑synuklein hade förändringar i mino liten effekt. Markörer för celldöd i den visuella hjärnregionen speglade detta mönster. Teamet observerade också en markant ansamling av fettdroppar — små sfärer för fettlagring — inte i neuronerna själva utan i närliggande gliaceller. Dessa droppar ökade med åldern i alpha‑synuklein‑flugor och påverkades av enzymer som bygger upp eller bryter ner triglycerider, vilket framhäver ett aktivt metabolt samspel mellan neuroner och glia under stress.
Metabol omkoppling och alpha‑synuklein‑klumpning
Metabolitmätningar från flughjärnor visade att alpha‑synuklein‑uttryck var kopplat till en flaskhals i cellens energi producerande cykel: citrat och isocitrat, två intermediärer i trikarboxylsyracykeln (TCA), ackumulerades kraftigt, medan efterföljande steg förändrades mer måttligt. Mjölksyranivåer steg också, i linje med ökad glykolys. Samtidigt visade detaljerad lipidprofilering förändringar i balansen av membranfosfolipider och deras fettsyresammansättning, med en förskjutning mot arter som är mer benägna att genomgå oxidativ skada. När teamet minskade flera GPAT‑enzymer — mino i mitokondrier och besläktade enzymer i det endoplasmatiska retikulumet — ackumulerades alpha‑synuklein fortfarande, men dess benägenhet att bilda högre ordningens oligomerer (flerproteinkomplex) minskade, och mitokondrier visade färre tecken på reaktivt syre‑stress och ”åldrande”.
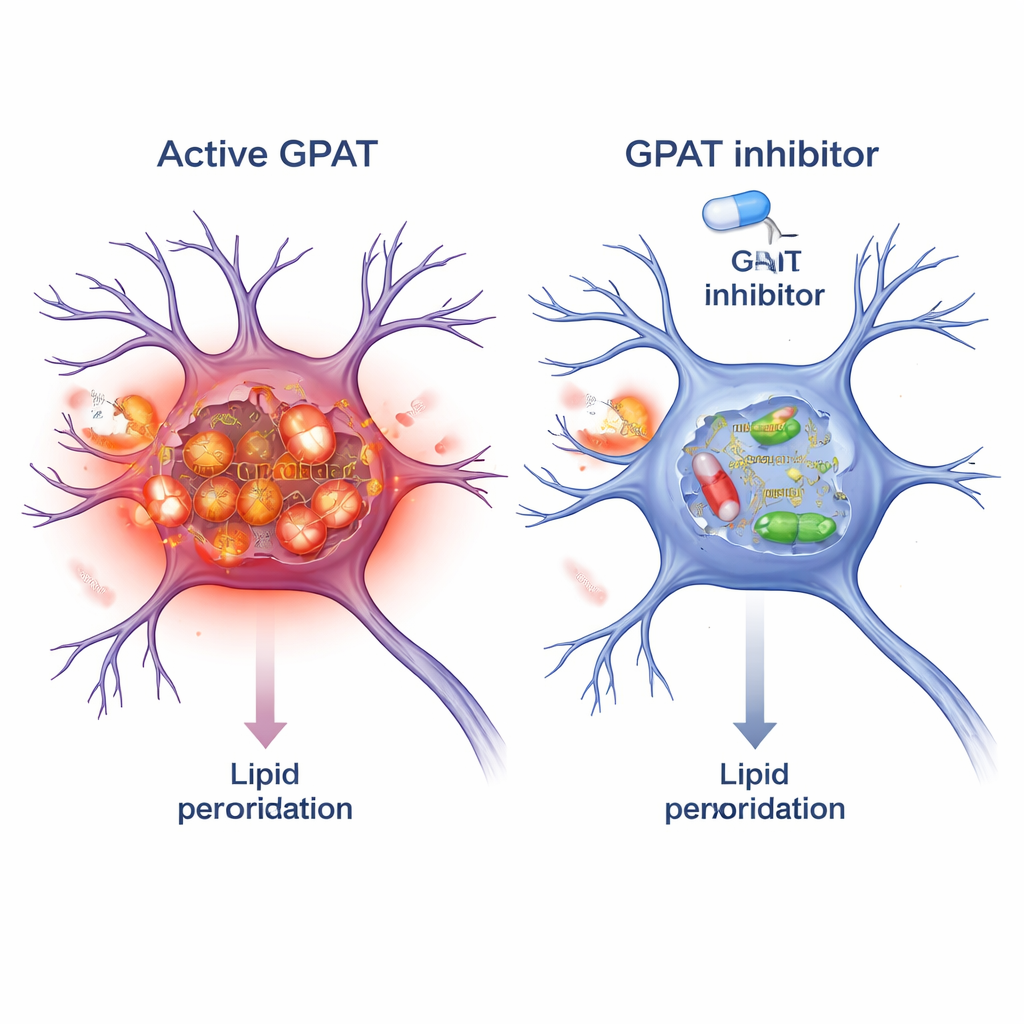
Att blockera GPAT som en skyddsstrategi
Eftersom GPAT är ett enzym kan det riktas med små molekylära läkemedel. Forskarna testade FSG67, en befintlig GPAT‑hämmare ursprungligen utvecklad för fetma och diabetes. I alpha‑synuklein‑flugor återgav tillsats av FSG67 i födan fördelarna av genetisk GPAT‑nedreglering: förbättrad rörelse, bättre överlevnad av dopaminneuroner, färre skadliga alpha‑synuklein‑oligomerer och minskad mitokondriell oxidativ stress. För att undersöka om detta också gällde däggdjur behandlade de odlade musneuroner med förformade alpha‑synuklein‑fibriller, som sårar toxisk aggregation. Sambehandling med FSG67 minskade ansamlingen av fosforylerat alpha‑synuklein och sänkte flera oberoende markörer för lipidperoxidation i dessa neuroner.
Vad detta betyder för personer med Parkinsons
Enkelt uttryckt visar detta arbete att hur hjärnan hanterar fetter kan skruva upp eller ner alpha‑synuklein‑toxiciteten. När GPAT är mycket aktiv byggs mer sårbara lipider in i membran och lagringsdroppar, vilket gör dem lättare att oxidera; denna skadade fettrikedom tycks gynna skadliga former av alpha‑synuklein och belasta mitokondrierna, cellens kraftverk. Att dämpa GPAT — antingen genetiskt eller med ett läkemedel — förskjuter balansen mot mindre lipid“rost”, färre toxiska proteinkomplex och friskare neuroner. Även om dessa fynd är tidiga och kommer från flugor och odlade musceller, lyfter de fram lipidmetabolism, och GPAT i synnerhet, som en lovande ny vinkel för Parkinson‑terapier som kan komplettera strategier som direkt riktar sig mot alpha‑synuklein.
Citering: Ren, M., Lim, G.G.Y., Tang, W. et al. Glycerol 3-phosphate acyltransferase exacerbates α-synuclein-induced toxicity by increasing lipid peroxidation. Nat Commun 17, 1618 (2026). https://doi.org/10.1038/s41467-026-68325-3
Nyckelord: Parkinsons sjukdom, alpha-synuklein, lipidperoxidation, GPAT-hämmare, neurodegeneration