Clear Sky Science · sv
Kombinera spatial transkriptomik med vävnadsmorfologi
Att betrakta vävnader på två olika sätt
Läkare och forskare vill i allt högre grad inte bara veta vilka gener som är aktiva i en vävnad, utan också exakt var de slås på. Samtidigt fångar sjukhusens mikroskop redan rika bilder av vävnadsstrukturen som patologer använder dagligen. Denna artikel förklarar hur ett nytt fält försöker koppla ihop dessa två perspektiv — detaljerade kartor över genaktivitet och vanliga mikroskopbilder — och varför detta samspel kan leda till tidigare diagnoser, bättre cancergradering och djupare insikt i hur sjukdomar utvecklas och sprider sig.
Från utspridda celler till kartor över genaktivitet
I många år krävde kraftfulla ”omiks”-metoder att vävnader mals ned till en blandning av enstaka celler, vilket förstörde information om var varje cell kom ifrån. Spatial transkriptomik förändrade detta genom att mäta genaktivitet samtidigt som varje cells position i vävnaden bevaras. Resultatet är ett rutnät av punkter, där varje punkt har en profil över genuttryck och exakta koordinater. Självt ger dessa spatiala gendata redan nya insikter om cellulär mångfald och sjukdomsarkitektur. Men de förändras inte efter mätningen, och att upprepa experimentet är kostsamt. I kontrast är vävnadsbilder färgade med standardfärgningar, såsom den allmänt använda hematoxylin‑eosin (H&E), billiga och rikligt förekommande, och innehåller visuella ledtrådar om cellform, täthet och vävnadsorganisation.
Två sätt att förena bilder och gener
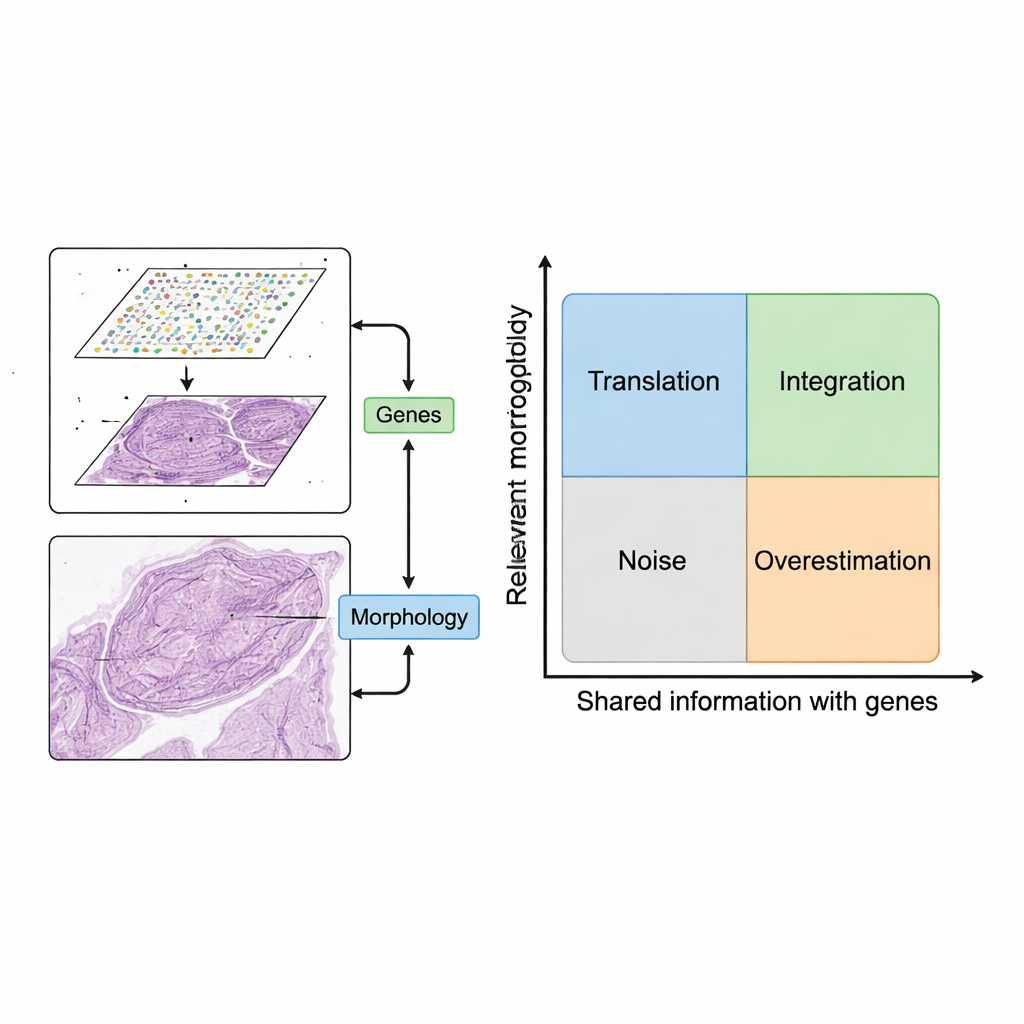
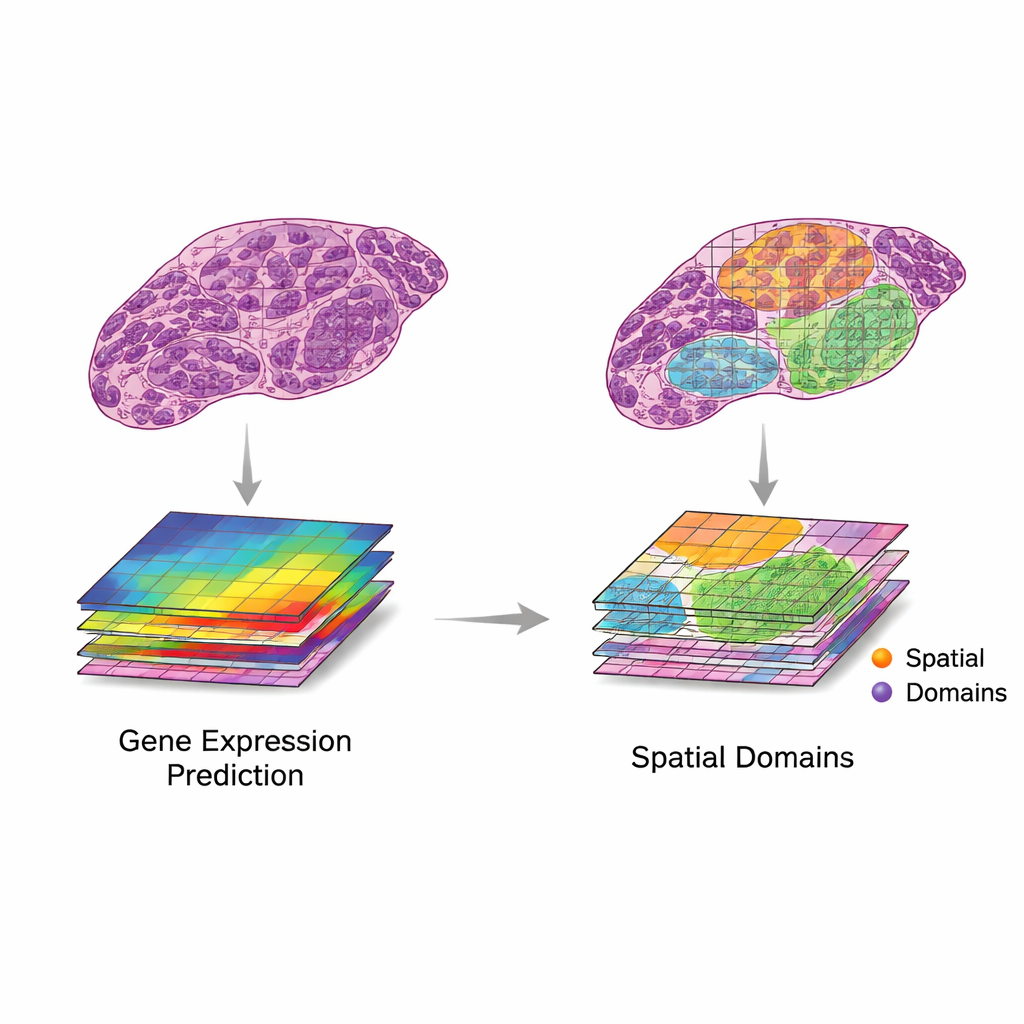
Översikten föreslår ett enkelt men kraftfullt ramverk för att använda dessa två datakällor tillsammans. Först paras bildpatchar ihop med närliggande punkter för genuttryck. Därefter extraherar datormodeller egenskaper från bilder — mönster som fångar form, textur och organisation — och jämför dem med mönster i genuttrycket. Författarna beskriver två önskvärda scenarier. I ”translation” följer bildegenskaper noggrant relevant genaktivitet, vilket gör det möjligt för modeller att förutsäga vilka gener som är aktiva enbart från vävnadsbilden. Detta kan användas för att fylla igen saknade genmätningar, nå högre upplösning än ursprungsrutnätet eller härleda genaktivitet från rutinmässiga kliniska snitt utan extra laboratoriearbete. I ”integration” fångar bildegenskaper användbar information som gendata missar, såsom långsamma strukturella förändringar eller subtil vävnadsorganisation, vilket hjälper till att definiera tydligare regioner eller ”domäner” inom en vävnad.
När extra information hjälper — och när den stjälper
Inte varje bildegenskap är värd att använda. Författarna introducerar en konceptuell karta med två axlar: hur relevant en bildegenskap är för den biologiska frågan, och hur mycket den överlappar med geninformationen. Egenskaper som varken är relevanta eller relaterade till gener utgör brus, såsom färgningsartefakter. Egenskaper som följer genmönster men kopplas till obetydliga gener (som grundläggande housekeeping‑gener) kan få modeller att se bra ut på papper men bidra lite kliniskt värde. Genom att organisera metoder i fyra kvadranter — translation, integration, brus och överskattning — klargör ramverket när kombinationen av bilder och gener verkligen tillför insikt, och när den bara upprepar eller döljer vad som redan är känt.
Nuvarande verktyg, tester och växande smärtpunkter
En snabbvuxande våg av artificiella intelligensmetoder försöker nu utföra translation och integration på verkliga data. Tidiga system byggde på konvolutionella neurala nätverk, medan nyare använder transformermodeller, grafneurala nätverk och multiskaliga modeller som kan ta in detaljer från små cellstrukturer upp till helbildskontext. Dessa metoder har använts för att förutsäga genaktivitet från H&E‑bilder, generera superupplösta kartor och hjälpa till att identifiera vävnadsregioner med distinkt beteende. För att bedöma prestanda förlitar sig forskare på statistiska mått som korrelation mellan förutsagda och observerade gennivåer, eller överensstämmelse mellan AI‑definierade regioner och expertpatologers etiketter. Dataset är dock fortfarande små och varierade, och jämförelser mellan studier är svåra. Många rapporterade förbättringar kan spegla överanpassning eller framgång på gener och mönster som har ringa klinisk betydelse.
Vart detta kan leda
Författarna avslutar med att kombinera spatiala genkartor med vävnadsbilder är lovande men fortfarande i ett tidigt skede. Dagens modeller når ofta bara måttlig noggrannhet och har ännu inte visat sig redo för rutinmässig medicinsk användning. Framtida framsteg kommer sannolikt från bättre bildegenskaper, särskilt stora ”foundation‑modeller” tränade på miljontals patologislide‑bilder, och från att fokusera på gener och mönster som verkligen påverkar patientvården. Noggrant utformad integration skulle en dag kunna avslöja tidiga varningstecken på sjukdom genom att upptäcka diskrepans mellan hur vävnaden ser ut nu och vad dess gener förutspår kommer att hända framöver. Kort sagt lägger detta arbete fram en färdplan för att omvandla rutinmässiga mikroskopbilder till rika, geninformella kartor som hjälper läkare att förstå och behandla sjukdomar mer precist.
Citering: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Nyckelord: spatial transkriptomik, vävnadsmorfologi, digital patologi, prediktion av genuttryck, bild‑AI