Clear Sky Science · sv
ATGL gör hepatocellulära carcinomceller känsligare för genotoxiska läkemedel genom att modulera p53:s acetylerings-/fosforyleringsstatus
Att förvandla fettnedbrytning till en cancersvaghet
Standardkemoterapi mot levercancer misslyckas ofta eftersom tumörceller är ovanligt skickliga på att överleva DNA‑skador. Denna studie utforskar en oväntad allierad inne i just dessa cancerceller: ett enzym som bryter ner lagrat fett. Genom att öka mängden av detta enzym, kallat ATGL, fann forskarna att de kunde pressa levertumörceller att sluta laga sina DNA‑skador och istället gå i självdöd. Arbetet blottlägger en dold koppling mellan hur cancerceller hanterar fett och hur de svarar på kraftfulla DNA‑skadande läkemedel, vilket pekar på nya sätt att få befintliga behandlingar att fungera bättre.
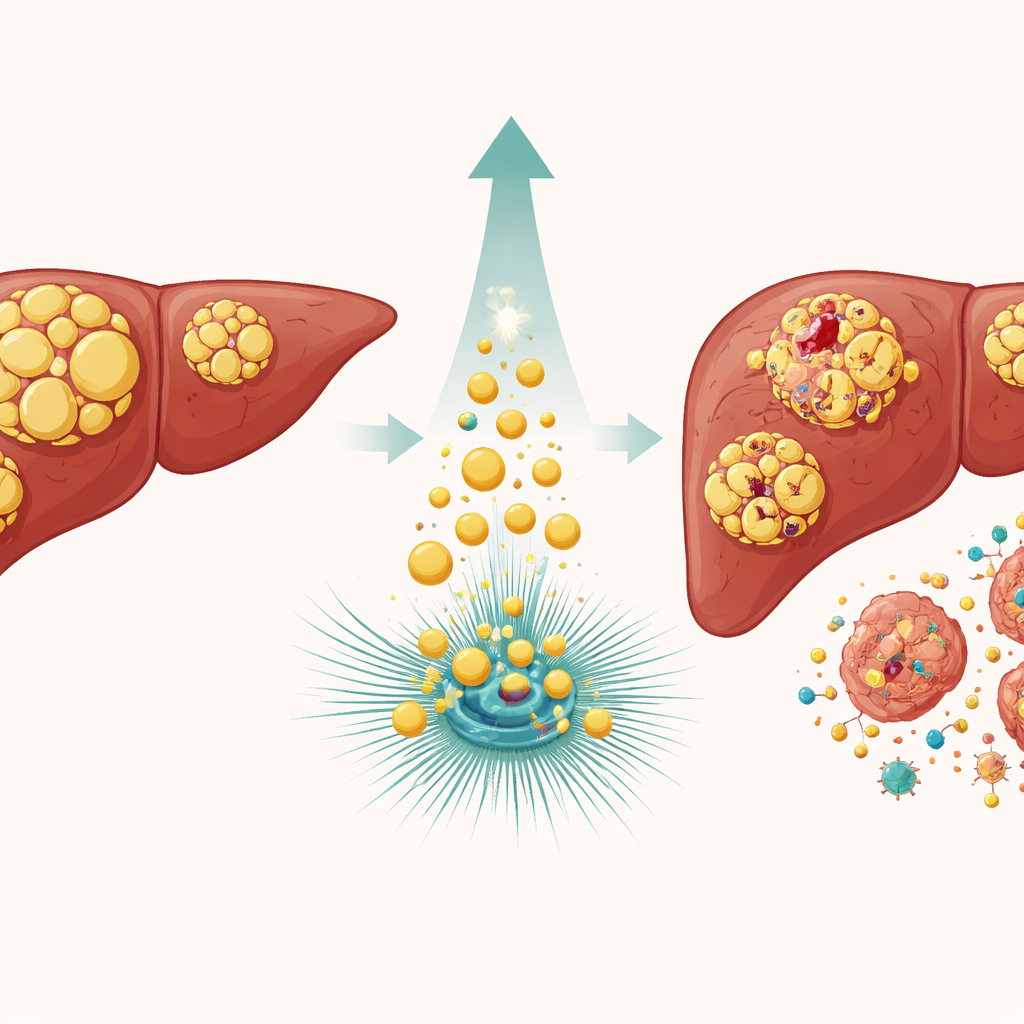
Varför levertumörer motstår tuffa läkemedel
Levercancer, särskilt hepatocellulärt carcinom, är en av de vanligaste och mest dödliga tumörtyperna i världen. Många patienter får läkemedel som skadar DNA, såsom etoposid och doxorubicin, i hopp om att tvinga cancercellerna in i en dödlig kris. Ändå undkommer dessa celler ofta genom att pausa sin tillväxt och aktivera reparationssystem som styrs av ett väktarprotein känt som p53. Om skadan kan lagas återupptar cellerna delningen; om inte kan p53 också utlösa programmerad celldöd. Den centrala gåtan är vad som får p53 att luta åt reparation kontra självdöd, och varför vissa tumörer förblir så segt resistenta mot terapi.
Ett fett‑skärande enzym tippar balansen
Teamet fokuserade på ATGL, ett enzym som beskär lagrade fetter i små cellulära reservoarer kallade lipidkroppar. I levercancer är ATGL‑nivåerna vanligen lägre än i frisk vävnad, och tidigare arbete antydde att tvångsmässig överproduktion av ATGL saktade tumörtillväxt. Här konstruerade forskarna levercancercellinjer för att antingen överproducera ATGL eller minska dess nivå, och utsatte dem sedan för DNA‑skadande läkemedel. Celler med extra ATGL ackumulerade betydligt fler tecken på brutet DNA, medan celler med reducerat ATGL uppvisade mindre skada. Att blockera ATGL:s klyvningsförmåga med en specifik hämmare, eller att uttrycka en mutant form som inte fungerar, utplånade denna förhöjda känslighet, vilket visar att enzymets fettbrytande aktivitet i sig är avgörande.
Omprogrammering av cellens beslut: reparera eller dö
Vidaregranskning visade att forskarna undersökte p53, som beter sig som en molekylär trafikpolis efter DNA‑skada. p53:s agerande styrs av små kemiska märken som läggs på specifika positioner. I ATGL‑rika celler orsakade genotoxiska läkemedel att p53 fick fler av en typ märkning (acetyleringsgrupper) och relativt färre av en annan (fosfatgrupper). Denna förskjutning gynnade aktivering av gener som främjar celldöd, såsom Puma, samtidigt som gener som p21 och GADD45 som normalt pausar cellcykeln och stödjer DNA‑reparation dämpades. Som ett resultat, även efter att läkemedlet spolats bort, lyckades inte ATGL‑höga celler bli av med markörer för DNA‑skada utan gick mot apoptos istället för återhämtning.
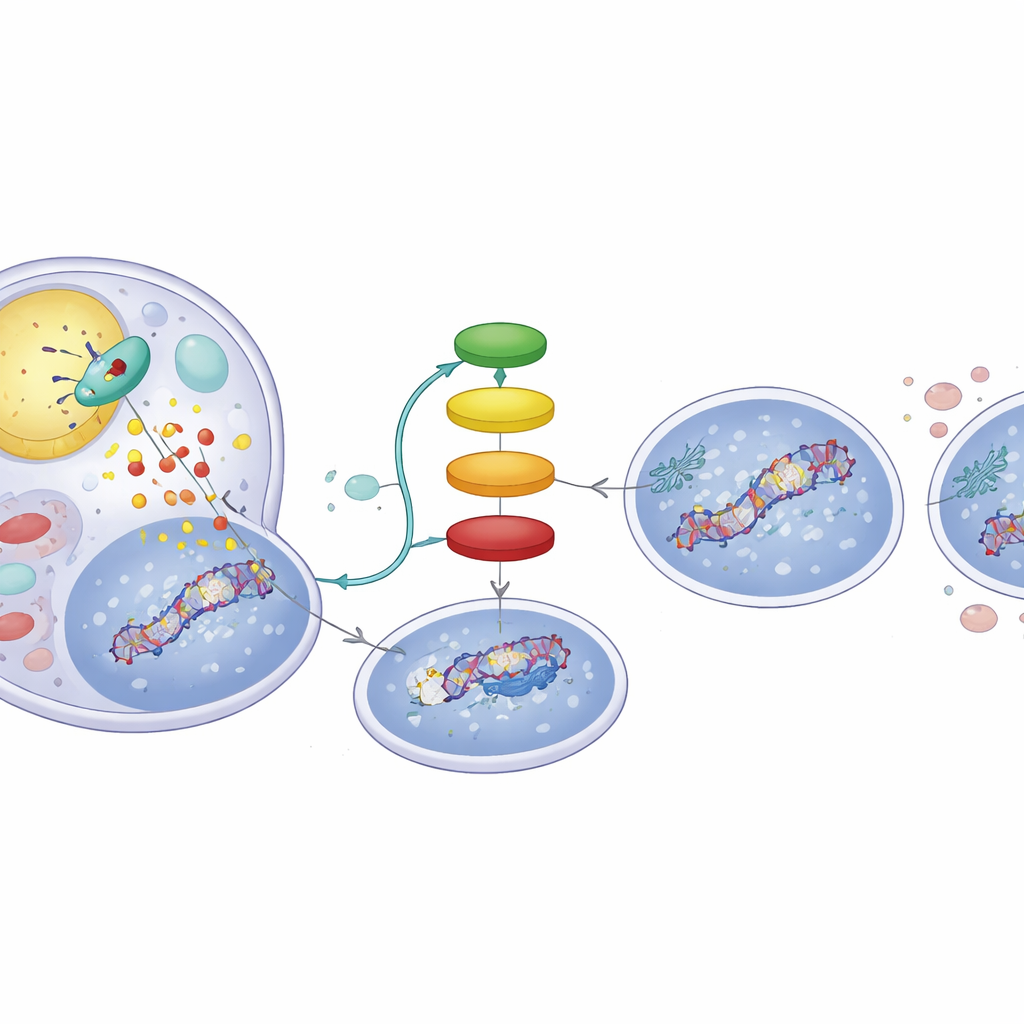
En fettdriven signalkedja inne i tumörceller
Hur ändrar fettnedbrytning p53:s märkningar? Nedbrytningsprodukterna från ATGL:s aktivitet är fria fettsyror som kan fungera som budbärare. Studien visar att dessa fettsyror aktiverar en nukleär receptor kallad PPARα, som i sin tur ökar aktiviteten hos p300, ett protein som fäster acetylmärken på p53. När forskarna använde en PPARα‑aktiverande förening reproducerade de beteendet hos ATGL‑rika celler: ökade DNA‑skadesignaler och en p53‑profil som lutade mot apoptos. Omvänt utplånade blockering av p300 de ATGL‑inducerade förändringarna i p53 och minskade DNA‑skadan, vilket betonar att en ATGL → PPARα → p300‑kedja är central för denna omkoppling. Analyser av hundratals mänskliga levertumörer från publika dataset bekräftade denna koppling och visade att tumörer med högre ATGL‑uttryck också tenderar att uppvisa starkare PPARα‑ och p300‑signaturer samt uttryck av p53‑reglerade gener.
Vad detta kan innebära för framtida behandlingar
Enkelt uttryckt visar studien att när levercancerceller uppmuntras att bryta ner lagrat fett via ATGL blir de mindre benägna att reparera kemoterapiinducerat DNA‑skada och mer benägna att genomgå ordnad celldöd. Det föreslår två praktiska möjligheter: att mäta ATGL‑nivåer kan hjälpa till att förutsäga vilka patienters tumörer som svarar bättre på genotoxiska läkemedel, och att öka ATGL‑aktivitet eller dess nedströms PPARα‑väg kan användas tillsammans med befintliga kemoterapier för att övervinna resistens. Medan ytterligare tester i djurmodeller och patienter behövs, lyfter arbetet fram ett slagkraftigt budskap: i levercancer kan det att göra tumörceller "magrare" på mikroskopisk nivå också göra dem mer sårbara för livsavgörande läkemedel.
Citering: Castelli, S., De Cristofaro, A., Desideri, E. et al. ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status. Cell Death Discov. 12, 164 (2026). https://doi.org/10.1038/s41420-026-03048-4
Nyckelord: hepatocellulärt carcinom, ATGL, DNA-skaderespons, p53-signalering, lipidmetabolism vid cancer