Clear Sky Science · sv
Metyleringstillståndet hos proteinfosfatas 2A påverkar α-synukleinopati i musmodeller
Varför detta spelar roll för hjärnhälsan
Parkinsons sjukdom och närliggande sjukdomar berövar människor gradvis rörlighet, minne och självständighet. En huvudmisstänkt är ett hjärnprotein kallat alfa-synuklein som kan veckas fel, klumpa ihop sig och skada nervceller. Denna studie ställer en hoppfull fråga: i stället för att angripa proteinet direkt, kan vi finjustera hjärnans egna städsystem för att förhindra att alfa-synuklein blir giftigt?
Berättelsen om ett klibbigt protein
Vid Parkinsons sjukdom och demens med Lewy-kroppar ansamlas vridna klumpar av alfa-synuklein inne i nervceller och bildar de klassiska ”Lewy-kropparna.” En specifik kemisk märkning på detta protein, tillsatt vid en plats kallad serin 129, är starkt kopplad till dess mest skadliga form. När denna märkning är riklig är alfa-synuklein mer benäget att bilda styva fibriller och aggregat. Hjärnan balanserar normalt sådana märkningar med enzymer som lägger till dem och andra som tar bort dem. Eftersom många enzymer kan lägga till märkningen är det osannolikt att blockera bara ett av dem räcker. I stället fokuserade författarna på huvudfamiljen av enzymer som tar bort märkningen, kallad proteinfosfatas 2A, eller PP2A, som fungerar som ett molekylärt suddgummi för denna farliga modifikation.
Hjärnans suddgummi och dess två strömbrytare
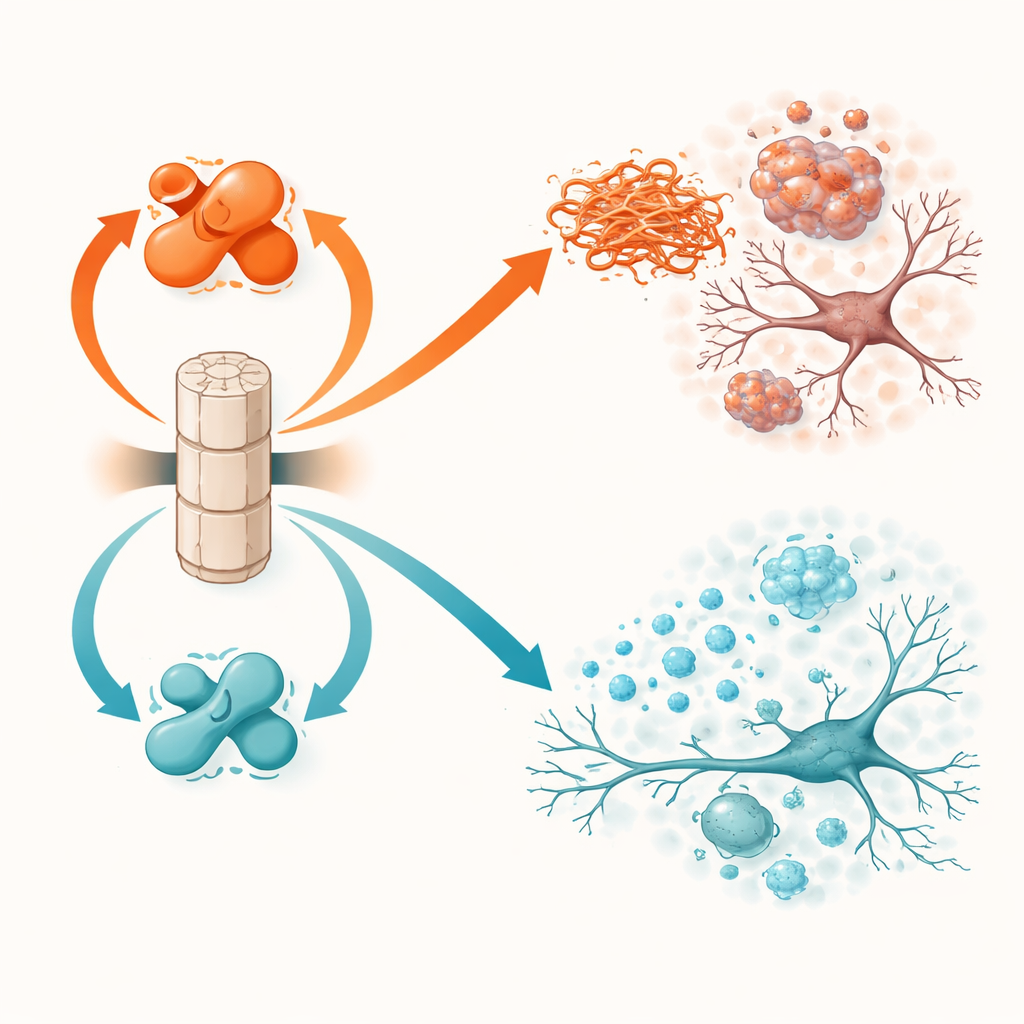
PP2A arbetar inte på full styrka som standard. Dess aktivitet beror på en liten kemisk märkning, kallad metylering, på en av dess subenheter. Två andra proteiner styr denna brytare: LCMT-1 lägger till märkningen och vänder PP2A mot en mer aktiv, skyddande form, medan PME-1 tar bort den och skjuter PP2A mot en mindre aktiv, skadlig form. Tidigare arbete i mänsklig hjärnvävnad visade att LCMT-1 tenderar att vara lägre och PME-1 högre vid Parkinsons och Lewy-body-demens, vilket lämnar PP2A underpresterande. Den aktuella studien testar direkt vad som händer när dessa brytare medvetet förskjuts åt endera håll i levande möss som utvecklar alfa-synukleinproblem.
Testa balansen i levande hjärnor
Forskarna använde två kompletterande musmodeller. I den ena var möss konstruerade för att producera mänskligt alfa-synuklein i hela hjärnan och utvecklade gradvis proteinklumpar samt rörelse- och minnesproblem med åldern. Dessa djur modifierades ytterligare för att överproducera antingen PME-1 (PP2A:s ”av”-brytare) eller LCMT-1 (PP2A:s ”på”-brytare) i framhjärnans neuroner. I den andra modellen injicerade teamet förformade alfa-synukleinfibriller i striatum, en djup hjärnregion involverad i rörelse. Dessa fibriller fungerar som frön som rekryterar normalt alfa-synuklein och sprider patologi över månader i i övrigt normala eller enzymändrade möss. I båda modellerna mätte forskarna proteinansamling, nervcellernas hälsa, hjärnans inflammation och beteende.
När suddgummit sviktar sprider sig skadan
Möss som överproducerade PME-1, och därmed hade mindre aktivt PP2A, klarade sig sämre. Hos alfa-synukleintransgena djur ledde ökad PME-1 till mer kraftigt märkt och aggregerat alfa-synuklein i cortex och hippocampus, större förlust av nervcellsstruktur, svagare neurala aktivitetsignaler och starkare aktivering av immunceller i hjärnan. Dessa förändringar översattes till sämre prestationer i rörelsetester och uppgifter för inlärning och minne. I fibrillinjektionsmodellen tillät PME-1-överuttryck att giftiga alfa-synukleinassemblys ansamlades och spreds mer omfattande, särskilt till de dopaminproducerande nervcellerna i substantia nigra, en nyckelregion som går förlorad vid Parkinsons sjukdom. Dessa möss visade mer uttalad förlust av dopaminfibrer, intensivare inflammation och större motoriska och boningsrelaterade (nesting) brister.
Sätta på suddgummit igen
Den motsatta manipulationen, att överproducera LCMT-1 för att hålla PP2A starkt metylerat och aktivt, hade i stort sett skyddande effekter. Hos alfa-synukleintransgena möss minskade LCMT-1 mängden märkt, aggregerat protein till nära normala nivåer och bevarade både neuroners struktur och aktivitet. Inflammatoriska markörer var lägre och djuren presterade närmare friska kontroller i balans- och minnestester. I fibrill-seedningsmodellen begränsade LCMT-1 både lokal ansamling och långväga spridning av giftigt alfa-synuklein, skonsade dopaminneuroner från degeneration, reducerade mikroglial aktivering och dämpade nedgången i motorisk koordinering och nestingbeteende. Genomgående översattes förskjutningen av PP2A mot dess aktiva, metylerade tillstånd konsekvent från molekylära förbättringar till funktionellt skydd.
Vad detta kan innebära för framtida behandlingar
För icke-specialister är budskapet enkelt: hjärnan har ett inbyggt suddgummi som kan ta bort en skadlig märkning på alfa-synuklein och förhindra att det förvandlas till farliga klumpar. När detta suddgummi försvagas blir skada, inflammation och symtom värre; när det stärks skyddas nervcellerna. Studien ger direkt bevis i levande djur för att metyleringstillståndet hos PP2A är en huvudkontroll för alfa-synukleins toxicitet och dess följder. Detta pekar mot en ny terapeutisk strategi: i stället för att jaga varje skadlig form av proteinet kan läkemedel utformas för att putta PP2A och dess regulatorer LCMT-1 och PME-1 mot en mer skyddande inställning. Sådana angreppssätt kommer att kräva noggranna säkerhetstester, men de har potential att bromsa eller förebygga Parkinsons sjukdom och relaterade tillstånd genom att återställa hjärnans egen förmåga att hålla alfa-synuklein i schack.
Citering: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
Nyckelord: Parkinsons sjukdom, alfa-synuklein, proteinfosfatas 2A, neurodegeneration, hjärnans inflammation