Clear Sky Science · sv
Modulering av metabola signaturer för att mildra cabozantinib‑resistens i FLT3‑ITD akut myeloisk leukemi‑cellmodeller
Varför detta är viktigt för cancerbehandling
Många moderna cancerläkemedel är utformade för att rikta in sig på ett enda felaktigt protein i tumörceller. Dessa målinriktade läkemedel kan orsaka dramatiska remissioner, men cancer kan ofta anpassa sig och återväxa. Denna studie undersöker hur en form av blodcancer, akut myeloisk leukemi (AML), blir resistent mot ett sådant målinriktat läkemedel, cabozantinib, och hur omkoppling av cancercellernas energianvändning kan hjälpa läkare att kringgå den resistensen.
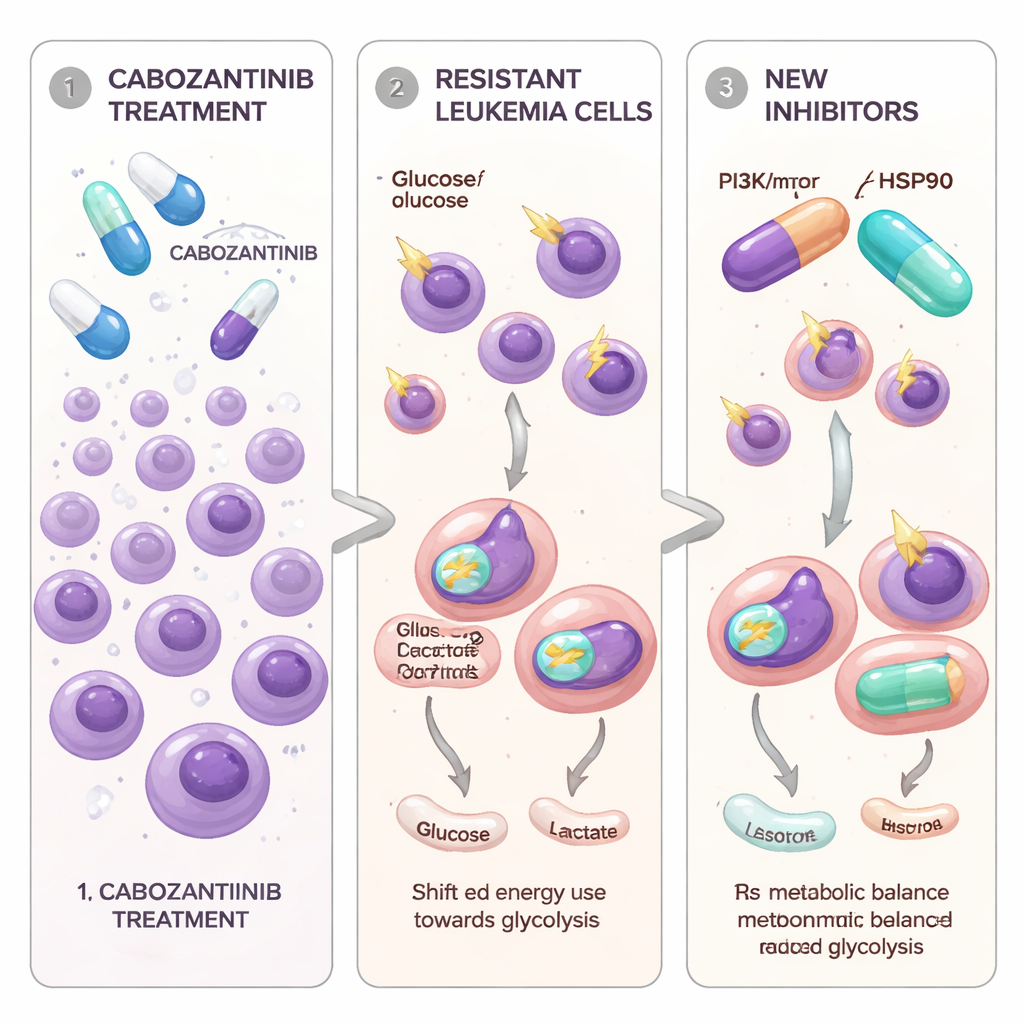
Leukemiceller som lär sig undvika ett målinriktat läkemedel
Forskarna fokuserade på AML‑celler som bär en mutation i en tillväxtsignalsbrytare kallad FLT3‑ITD, som är känd för att driva särskilt aggressiv sjukdom. Cabozantinib, en tablett som redan används för flera solida tumörer, kan starkt blockera FLT3‑drivna leukemiceller i laboratoriet. För att modellera vad som händer hos patienter över tid utsatte teamet två FLT3‑mutanta AML‑cellinjer för gradvis ökande doser av cabozantinib tills vissa celler överlevde och började växa igen. Dessa nya cellpopulationer, kallade Molm13‑XR och MV4‑11‑XR, kunde tolerera cabozantinibkoncentrationer många gånger högre än sina ursprungliga ”moder”‑celler. De blev också mindre känsliga för två andra godkända FLT3‑riktade läkemedel, sorafenib och quizartinib, samtidigt som de förblev sårbara för en annan hämmare, gilteritinib.
Genetiska ändringar som hjälper cancern att överleva
När forskarna granskade cellerna fann de att dessa läkemedelsresistenta leukemiceller bar nya förändringar i sitt FLT3‑gen. Båda resistenta linjerna hade förvärvat samma punktmutation, kallad D835Y, i en avgörande del av FLT3:s kinasdomän, ett känt hotspot för resistens mot flera läkemedel. En linje, MV4‑11‑XR, fick dessutom en ovanlig 1,3 kilobas‑deletion som tog bort ett helt exon i FLT3 och raderade en del av domänen som är viktig för läkemedelsbindning. Dessa förändringar verkade ha selekterats under långvarig exponering för cabozantinib: mutanta varianter av FLT3 blev mycket vanligare i de resistenta cellerna än i startpopulationen. Samtidigt var viktiga signalvägar nedströms om FLT3—såsom ERK, STAT5 och AKT—starkare aktiverade, vilket stödde snabbare tillväxt och större kolonibildning i de resistenta cellerna.
Cancerceller byter drivmedelssystem
Teamet undersökte sedan om resistensen var kopplad inte bara till genetik utan också till hur cellerna försörjer sig med energi. Med hjälp av RNA‑sekvensering och särskilda metabola tester fann de ett konsekvent mönster: cabozantinib‑resistenta celler förlitade sig mycket mer på glykolys—den snabba nedbrytningen av glukos i cytosolen—även när syre fanns tillgängligt. Dessa celler tog upp mer glukos, producerade mer laktat, uppvisade högre aktivitet av ett nyckelenzym kallat GAPDH och ökade uttrycket av flera gener kopplade till glykolys. I kontrast var cellernas mitokondrier — strukturerna som stödjer mer effektiv energiproduktion — mindre aktiva och mindre talrika. Mätningar av syreförbrukning visade att både basal och maximal mitokondriell respiration var nedsatt, och reaktiva syreföreningar inne i cellerna var förhöjda, vilket pekar på stressade, underpresterande mitokondrier.
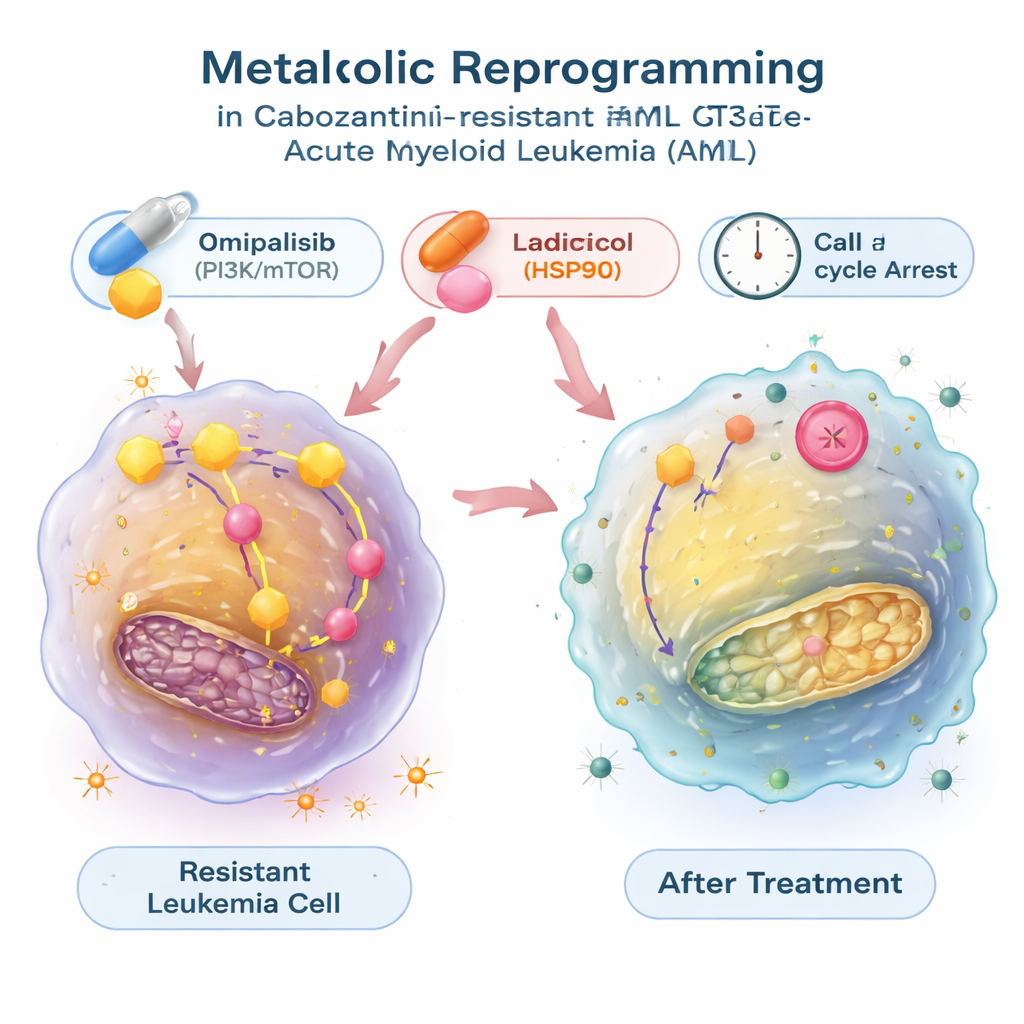
Hitta läkemedel som vrider tillbaka den metabola omkopplaren
För att se om denna energiförskjutning kunde vändas använde forskarna en stor offentlig databas som kopplar genuttrycksprofiler till effekterna av tusentals föreningar. De sökte efter läkemedel som förutspåddes motverka den metabola signaturen hos de resistenta leukemicellerna och fokuserade på två: radicicol, som blockerar ett proteinchaperon kallat HSP90, och omipalisib, som hämmar PI3K/mTOR‑signalvägen som kontrollerar tillväxt och metabolism. I laboratorietester bromsade båda molekylerna inte bara tillväxten av resistenta celler utan minskade också deras överaktiva glykolys, normaliserade glukosupptag och laktatproduktion samt sänkte uttrycket av glykolysrelaterade gener. Dessa föreningar förde leukemicellerna in i en vilofas i cellcykeln, och i fallet med radicicol utlöste de även betydande programmerad celldöd. I kombination med cabozantinib fungerade omipalisib—och i en modell även radicicol—synergistiskt och gjorde de läkemedelsresistenta cellerna lättare att döda.
Vad detta betyder för framtida leukemibehandlingar
För icke‑specialister är budskapet att leukemiceller kan undkomma ett målinriktat läkemedel inte bara genom att mutera dess direkta mål, utan också genom att ändra hur de skapar och använder energi. Studien visar att cabozantinib‑resistenta AML‑celler antar en ”sockersmältnings” strategi samtidigt som deras mitokondrier förlorar kraft. Genom att slå mot de vägar som stöder denna omkopplade metabolism—genom läkemedel som omipalisib eller HSP90‑hämmare—kan det vara möjligt att återställa känsligheten för cabozantinib och liknande behandlingar. Även om dessa fynd kommer från cellmodeller snarare än patienter, antyder de att kombinationer av målinriktade cancerläkemedel och metabolt modifierande medel kan vara ett lovande sätt att fördröja eller övervinna resistens i FLT3‑mutant AML.
Citering: Fu, YH., Ng, K.M., Tseng, CY. et al. Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models. Cell Death Discov. 12, 98 (2026). https://doi.org/10.1038/s41420-026-02957-8
Nyckelord: akut myeloisk leukemi, läkemedelsresistens, FLT3‑mutation, cancermetabolism, cabozantinib