Clear Sky Science · sv
DNA-polymeras kappa stabiliserad av Ptbp2 interagerar med MRE11 och främjar genomisk instabilitet i leukemi
Hur leukemiceller behåller brutet DNA och ändå överlever
Vårt DNA utsätts ständigt för angrepp, men friska celler är vanligen mycket skickliga på att hitta och åtgärda skador. I leukemi lär sig dock vissa celler att leva med brutet, instabilt DNA — och de vänder till och med den instabiliteten till en överlevnadsfördel. Denna studie avslöjar ett molekylärt ”samarbete” mellan ett splitsningsprotein (Ptbp2), ett särskilt DNA-kopierande enzym (DNA-polymeras kappa, eller Polk) och en skadesensor (MRE11) som hjälper leukemiceller att reparera tillräckligt med skador för att överleva, samtidigt som de ackumulerar den genetiska oordning som driver cancerutveckling.
En dold hjälpare i leukemiceller
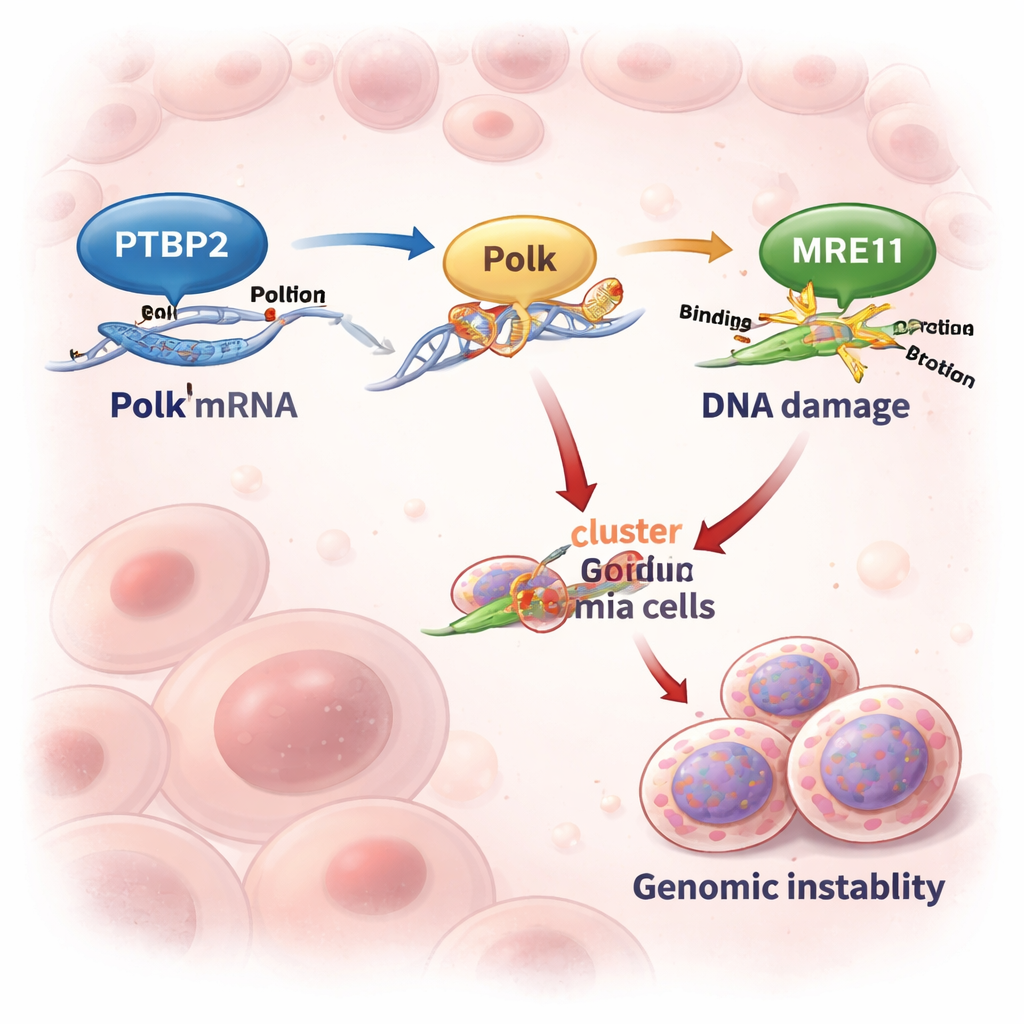
Forskarna fokuserade på kronisk myeloid leukemi (CML), en blodcancer som vanligtvis drivs av BCR::ABL1-fusionsgenen. Medan moderna läkemedel som blockerar BCR::ABL1 fungerar väl i tidig sjukdom reagerar många patienter i den aggressiva ”blastkris”-fasen dåligt. Tidigare arbete visade att Ptbp2, ett protein som binder RNA och påverkar hur budskapen bearbetas, ökas av BCR::ABL1 och agerar som ett onkogen i CML. Här upptäckte teamet att Ptbp2 fäster vid 3′ UTR i Polk-messenger-RNA och skyddar det från nedbrytning. Som ett resultat producerar leukemiceller mer Polk-protein när Ptbp2-nivåerna är höga.
Att slå på en felbenägen DNA-kopieringsmaskin
Polk är en ”reserv” DNA-polymeras som kan kopiera över skadat DNA när den vanliga replikeringsmaskineriet stannar upp. Denna förmåga kan rädda stressade celler — men det har sitt pris, eftersom Polk är felbenägen och kan införa mutationer. I cellinjer och patientprover från avancerad CML steg och sjönk nivåerna av Ptbp2 och Polk i takt med varandra. När forskarna slog ut Ptbp2 i leukemiceller sjönk Polk-nivåerna kraftigt och Polk-RNA:et degraderades nästan dubbelt så snabbt. Att återinföra Polk i Ptbp2-bristande celler återställde deras beteende, vilket visar att Ptbp2:s huvudroll här är att hålla Polk rikligt och aktivt.
Reparerar skador — men inte perfekt
För att se hur detta duo påverkar DNA-reparation behandlade forskarna celler med hydroxyurea, ett läkemedel som stoppar DNA-replikation och ofta används hos CML-patienter. Celler utan Ptbp2 drabbades av mycket mer DNA-skador, synliga som långa ”komet-svansar” och starka γH2AX-foci — kännetecken för brutna kromosomer. Dessa skadade celler hade större benägenhet att dö. I kontrast tålde celler med höga nivåer av Ptbp2 och Polk läkemedlet bättre, reparerade skador mer effektivt och överlevde, även om deras reparation var slarvig. Överuttryck av Polk i Ptbp2-knockout-celler räddade denna sårbarhet och bekräftade att Ptbp2–Polk-partnerskapet hjälper leukemiceller att klara replikeringsstress och undvika apoptos.
Ett DNA-skadenätverk som gynnar instabilitet
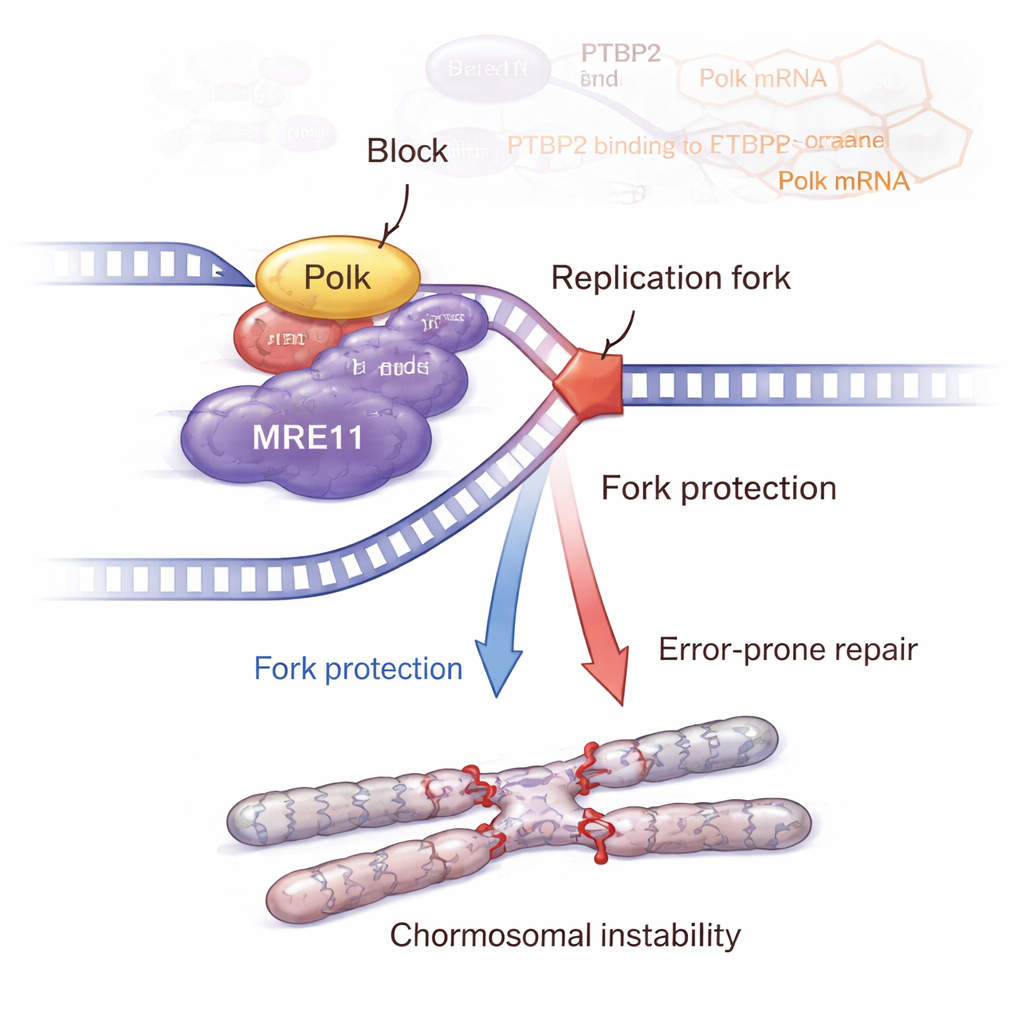
Berättelsen slutar inte med Polk. Teamet visade att Polk fysiskt interagerar med MRE11, en nyckelkomponent i MRN-komplexet som känner av DNA-brott och aktiverar ATM–CHK2-skaderesponsen. När Ptbp2 togs bort sjönk Polk, MRE11-nivåer och aktivitet minskade, och ATM–CHK2-signalerna försvagades. Att återföra Polk återställde MRE11 och dess aktivering. Detaljerade DNA-fiberexperiment visade att Ptbp2 och Polk hjälper till att skydda stoppade replikationsgafflar från att backas tillbaka, till stor del via MRE11. Att blockera MRE11 med ett läkemedel underminerade detta gaffelskydd och ökade DNA-skador. Paradoxalt nog samlade celler med aktiv Ptbp2–Polk–MRE11-signalering på sig fler kromosomala avvikelser, såsom sister chromatid exchanges, brott, glapp, multipolära spindlar och flerkärniga jätteceller — klassiska tecken på genomisk instabilitet som kan driva mer aggressiv cancer.
Från möss till möjliga nya behandlingar
I musemodeller gav leukemiceller med intakt Ptbp2 större, mer onormala tumörer än celler utan Ptbp2. Vävnader från dessa möss visade högre nivåer av Ptbp2, Polk, proliferationsmarkören Ki-67 och förvrängda strukturer vid celldelning. I en separat CML-lik modell driven av BCR::ABL1 gav tillskott av Ptbp2 ökat Polk och fler atypiska delande celler samt invasiva leukemikluster i mjälte och lever, vilket pekar på snabbare sjukdomsprogression. Tillsammans tyder dessa fynd på att Ptbp2–Polk–MRE11–ATM–CHK2-axeln tillåter leukemiceller att överleva intensiv DNA-stress samtidigt som de stadigt bygger upp skadliga mutationer.
Varför detta spelar roll för patienter
För en lekmannaläsare är huvudbudskapet att vissa leukemiceller undkommer kontroll genom att balansera på en knivsegg: de reparerar sitt DNA precis tillräckligt för att överleva, men inte så bra att de undviker mutationer. Ptbp2 stabiliserar Polk, som sedan samarbetar med MRE11 för att skydda stressat DNA och hålla igång skadesignaler — men denna reparation är ofullständig och främjar genetiskt kaos. Eftersom avancerad CML och andra cancerformer verkar bero på denna bräckliga balans kan riktade behandlingar mot Ptbp2 eller dess kontroll över Polk föra celler från överlevnad mot självförstörelse, vilket erbjuder en lovande ny infallsvinkel för terapi, särskilt i den svårbehandlade blastkris-stadiet.
Citering: Lama, S., Barik, B., IS, S. et al. DNA polymerase kappa stabilized by Ptbp2 interacts with MRE11 and promotes genomic instability in leukemia. Cell Death Discov. 12, 96 (2026). https://doi.org/10.1038/s41420-026-02951-0
Nyckelord: kronisk myeloid leukemi, genomisk instabilitet, DNA-reparation, DNA-polymeras kappa, PTBP2