Clear Sky Science · sv
Mitofagi vid pankreascancer: mekanistiska insikter och implikationer för nya terapeutiska strategier
Varför cellernas kraftverk spelar roll vid pankreascancer
Pankreasductalt adenokarcinom är en av de dödligaste cancerformerna, delvis därför att tumörer snabbt lär sig stå emot nästan alla behandlingar som används. Denna översiktsartikel undersöker en förvånansvärd aktör i den resistenshistorien: mitofagi, cellens inbyggda system för att identifiera och återvinna utslitna mitokondrier, de små ”kraftverken” som producerar energi. Att förstå hur pankreascancer kapar denna städprocess kan öppna nya vägar till effektivare och mer långvariga terapier.
Cellernas städpatruller och cancerns överlevnadstrick
Mitokondrier gör mycket mer än att generera energi; de hjälper till att styra metabolism, celldöd och hanteringen av skadliga molekyler kallade reaktiva syreradikaler. När mitokondrier skadas eller blir för många använder celler mitofagi för att märka och avlägsna dem. Detta kan ske via två huvudvägar. Den ena, känd som PINK1/Parkin‑vägen, bygger på en skadesensor (PINK1) och ett märkningsenzym (Parkin) för att flagga defekta mitokondrier för destruktion. Den andra använder receptorproteiner som BNIP3, NIX och FUNDC1 på mitokondriernas yta för att direkt koppla dem till cellens avfallsblåsor, autophagosomer, utan samma märkningssteg. Dessa vägar samarbetar ofta och ger celler flexibla sätt att anpassa sitt mitokondrieutbud till stress, som lågt syre eller läkemedelsexponering. 
Hur pankreastumörer omformar sin energianvändning
Pankreastumörer lever i en hård miljö: de är dåligt försörjda med blod och syre och omgivna av tät ärrlik vävnad. För att överleva kopplar cancerceller om sina energisystem, ofta genom att gå från syreberoende andning i mitokondrier till en mer primitiv sockerförbränning som kallas glykolys. Mitofagi hjälper till att finjustera denna förskjutning. I tidig tumörutveckling påskyndar förlust av PINK1 eller Parkin cancerbildning genom att tillåta järnansamling, ökad förekomst av reaktiva syreradikaler och en drivkraft mot glykolys. Samtidigt kan ett annat mitofagiprotein, NIX, hjälpa till att avlägsna ännu fungerande mitokondrier, vilket skjuter cellerna ytterligare mot glykolytisk metabolism och snabbare tillväxt. BNIP3 uppvisar däremot ett mer komplext mönster: det verkar tidigt, tystas senare i många avancerade tumörer och när det återställs kan det bromsa cancercellstillväxt, vilket tyder på att vissa mitofagivägar begränsar cancer medan andra driver den.
Cancerstamceller och tumörens omgivning
Alla cancerceller är inte likadana. En liten undergrupp, ofta kallad cancerstamceller, kan självförnya sig, ge upphov till nya tumörer och klara kemoterapi särskilt väl. I pankreascancer är dessa celler beroende av mitofagi för att förbli metaboliskt flexibla. Ett modifierande protein kallat ISG15 stödjer denna process och hjälper till att bevara deras stamliknande egenskaper, vilket kopplar mitofagi direkt till återfall och behandlingsmisslyckande. Utöver cancercellerna själva formar mitofagi även tumörmikromiljön — blandningen av stödjande celler, immunceller och bindväv runt tumören. Cancerassocierade fibroblaster kan exempelvis drivas in i en ”reverse Warburg‑effekt”, där de ökar sin egen mitofagi och glykolys och sedan matar tumören med energirika biprodukter. Mitofagi påverkar också hur immunceller känner igen och attackerar cancer, genom att ändra presentationen av immunflaggor som MHC‑I och bromsar som PD‑L1 som tumörer använder för att gömma sig.
Varför att blockera eller förstärka städningen kan ändra läkemedelssvar
Många standard- och experimentella behandlingar mot pankreascancer, inklusive cytostatikan gemcitabin och ett nytt riktat läkemedel mot den vanliga KRAS‑G12D‑mutationen, skadar mitokondrier. Tumörceller reagerar ofta genom att slå på mitofagi, rensa skadorna och undkomma död. Studier visar att PINK1‑driven mitofagi kan dämpa cytostatika och vissa naturföreningars dödande effekter, vilket hjälper cancerceller att överleva. Å andra sidan kan överdriven stimulering av mitofagi — särskilt via BNIP3 och NIX — under vissa förhållanden avkläda celler på för många fungerande mitokondrier, driva dem i en energikris och främja celldöd. Denna dubbla natur betyder att mitofagi antingen kan skydda tumörer från behandling eller, om den pressas på rätt sätt, göra dem mer sårbara. 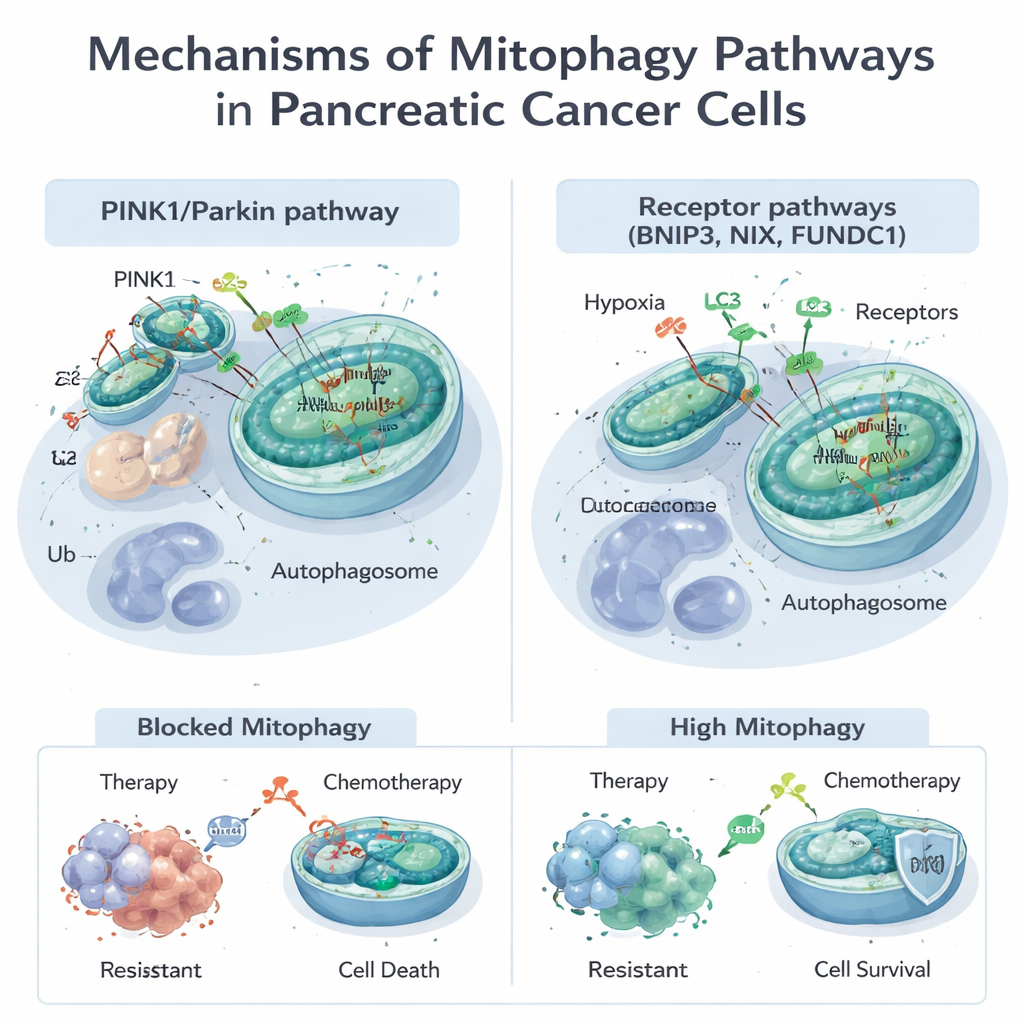
Nya behandlingsidéer och behovet av smart patientmatchning
Eftersom mitofagi ligger i skärningspunkten mellan metabolism, stressvar och celldöd är det ett attraktivt mål för kombinationsbehandlingar. Nuvarande läkemedelsstrategier verkar mest indirekt: vissa föreningar stör mitokondriernas delning och sammansmältning; andra, som klorokin och hydroklorokin, blockerar de sista stegen i återvinningsprocessen genom att förhindra sammansmältningen av avfallsblåsor med sura nedbrytningskompartment. Flera kliniska prövningar testar dessa läkemedel tillsammans med standardkemoterapi vid pankreascancer. Resultaten hittills är dock blandade — vissa patienter gynnas, andra gör det inte. En del av problemet är att tumörer skiljer sig kraftigt i sin grundläggande mitofagi‑ och metabola profil: vissa är starkt beroende av mitokondriell respiration, andra av glykolys. Författarna menar att framtida framgångar kommer att bero på bättre biomarkörer och gensekvenser för att gruppera patienter efter deras mitofagiaktivitet och energianvändning, samt på utveckling av mer precisa läkemedel som selektivt justerar specifika mitofagivägar i stället för att trubbigt slå på eller av autofagi. För lekmän är huvudbudskapet att kunna läsa och varsamt finjustera detta mitokondriella städsystem kan hjälpa till att omvandla pankreascancer från en envis fiende till en mer hanterbar sjukdom.
Citering: Wang, Z., Lyu, Z., Palmen, R. et al. Mitophagy in pancreatic cancer: mechanistic insights and implications for novel therapeutic strategies. Cell Death Discov. 12, 93 (2026). https://doi.org/10.1038/s41420-026-02948-9
Nyckelord: pankreascancer, mitofagi, mitokondrier, kemoterapiresistens, tumörmetabolism