Clear Sky Science · sv
Hämning av RRM1 gör lungadenokarcinom känsligare för behandling med decitabin
Från en ljummen medicin till en kraftfullare allierad
Lungcancer är fortfarande en av de mest dödliga cancerformerna, och många patienter får så småningom slut på effektiva behandlingsalternativ. Läkare har länge hoppats att läkemedel som försiktigt omprogrammerar cancercellernas DNA, istället för att bara förgifta delande celler, skulle kunna hjälpa. Ett sådant läkemedel, decitabin, fungerar väl vid blodcancer men har varit en besvikelse vid solida tumörer som lungcancer. Denna studie ställer en enkel, praktisk fråga med stora implikationer: finns det ett sätt att få lungtumörer att slutligen svara på decitabin, med verktyg vi redan förstår?
Varför ett beprövat läkemedel misslyckas i solida tumörer
Decitabin är en liknande variant av en av DNA:s byggstenar. När den införlivas i en cells DNA under kopiering kan den sudda ut onormala kemiska märken som tystar skyddande gener, inklusive tumörsuppressorer och immunrelaterade gener. I leukemier hjälper detta till att återställa celler mot ett friskare tillstånd. I lungtumörer fungerar läkemedlet däremot marginellt. Författarna misstänkte att problemet inte var vad decitabin gör, utan hur lite av det som faktiskt byggs in i DNA i celler från solida tumörer. Genom att mäta mycket små mängder läkemedel inbyggt i DNA över många cancercellinjer bekräftade de att celler som inkorporerade mer decitabin var mycket lättare att döda med läkemedlet.
En cellulär grindvakt som blockerar läkemedlet
För att ta reda på vad som begränsar läkemedlets inträde i DNA undersökte forskarna gener som är involverade i hanteringen av nukleosider — de råa materialen för DNA. Ett enzym, kallat RRM1, stack ut. RRM1 är en del av ett maskineri som omvandlar vanliga byggstenar till aktiva former som används för att bygga DNA. I lungadenokarcinom var detta enzym ovanligt rikt i tumörer jämfört med normalt lungvävnad, och patienter med lägre RRM1-nivåer tenderade att leva längre. Över en panel av cancercellinjer gick högre RRM1-nivåer hand i hand med lägre inkorporering av decitabin, vilket starkt tyder på att detta enzym fungerar som en grindvakt som tränger undan läkemedlet.
Avväpna grindvakten för att hjälpa läkemedlet att fungera
Teamet frågade sedan vad som händer om de delvis inaktiverar RRM1. Med genetiska verktyg sänkte de RRM1 i lungcancerceller utan att döda cellerna direkt. I sig hade denna reduktion bara en mild effekt på tillväxten. Men när den kombinerades med låga doser decitabin blev effekten dramatisk: kolonier av lungcancerceller krympte kraftigt i odlingsskålar och tumörer växte mycket långsammare i möss. Viktigt är att dessa effektiva doser var väl tolererade, utan uppenbara skador på blod, lever eller njurfunktion hos djuren. På molekylär nivå tillät blockad av RRM1 att mer decitabin byggdes in i DNA, vilket ledde till en starkare förlust av metyleringsenzymet DNMT1 och ett större fall i global DNA-metylering. Detta återaktiverade i sin tur tumörsuppressorgener som tidigare varit avstängda.
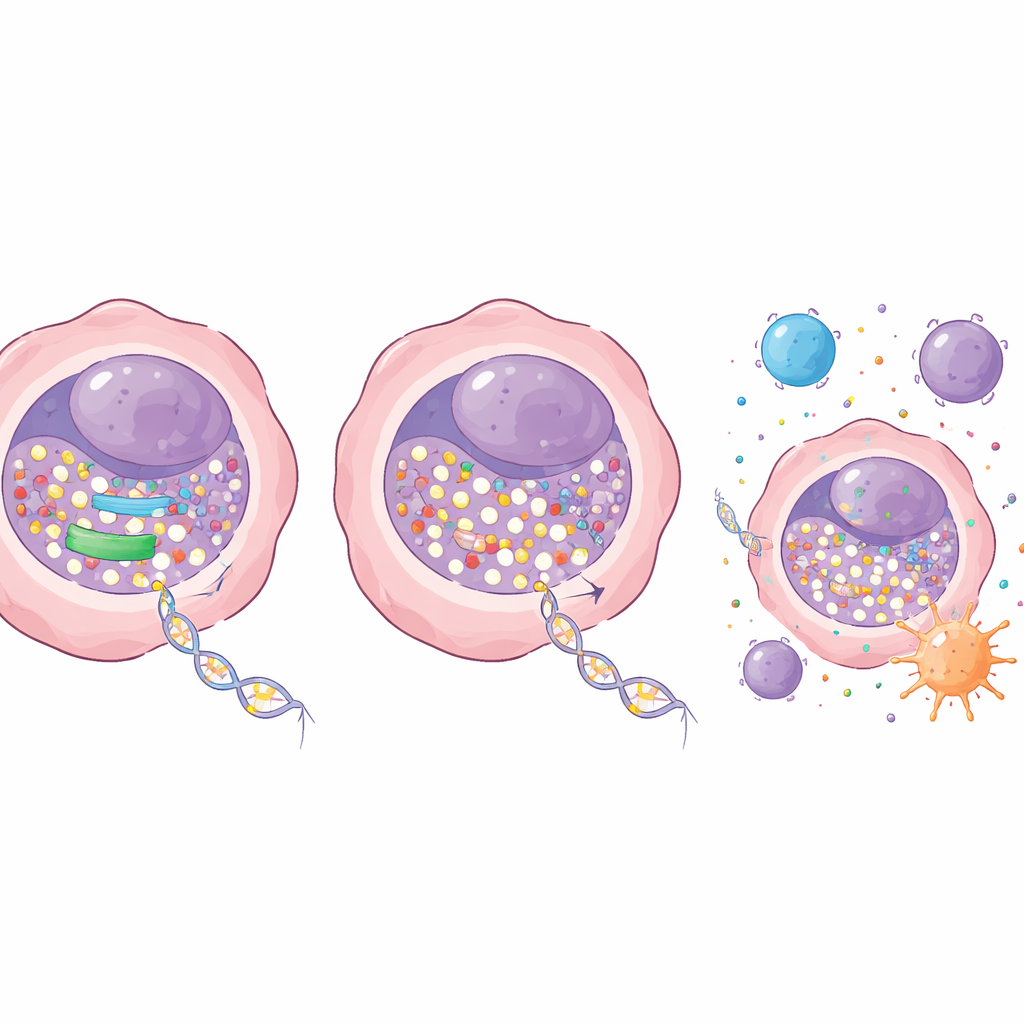
Sätta på immunalarmen inne i tumörerna
Utöver att bromsa celldelning förändrade kombinationsterapin hur cancerceller interagerar med immunsystemet. Mer decitabin i DNA ökade signaler om DNA-skador inne i cellerna och drev dem mot programmerad celldöd. Samtidigt förstärkte det aktiviteten i ett internt larmsystem centrerat kring STING-vägen, som känner av felplacerat DNA och utlöser antivirala liknande immunresponser. När RRM1 blockerades aktiverade decitabin denna väg och dess nedströms gener starkare, inklusive de som rekryterar och stimulerar immunceller. I musmodeller av lungcancer med intakta immunsystem gav kombinationen av decitabin och ett läkemedel som hämmar RRM1 starkare tumörkontroll än någon av behandlingarna ensam, utan ökad tydlig toxicitet. Författarna fann också att denna enzymblockerande strategi förbättrar decitabin specifikt, och kan faktiskt vara verkningslös eller negativ mot ett närbesläktat läkemedel, azacitidin, vilket understryker behovet av att matcha rätt partner.
Vad detta kan betyda för patienter
Sammantaget målar arbetet upp en tydlig bild: ett överaktivt DNA-byggnadsenzym i lungtumörer begränsar hur mycket decitabin når sitt mål. Genom att delvis blockera detta enzym tvingas cancerceller använda mer av läkemedlet istället för sina vanliga byggstenar. Denna förskjutning gör att låga doser decitabin effektivare kan återaktivera skyddande gener, skada cancercellernas DNA och väcka immunförsvaret — allt samtidigt som det förblev tolerabelt i djurmodeller. För patienter antyder detta en realistisk väg framåt: att återanvända eller förfina hämmare av RRM1-enzymet i kombination med lågdoserat decitabin och potentiellt med moderna immunoterapier, för att förvandla ett tidigare otillräckligt läkemedel till en användbar komponent i lungcancerbehandling.
Citering: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Nyckelord: lungadenokarcinom, decitabin, DNA-metylering, ribonukleotidreduktas, cancerimmunoterapi