Clear Sky Science · sv
ROR1-PI3K/AKT-signalering driver adaptiv resistens mot cellcykelblockad vid TP53‑muterad äggstockscancer
Varför denna forskning är viktig för kvinnors hälsa
Äggstockscancer är en av de dödligaste cancerformerna hos kvinnor, till stor del eftersom tumörer ofta utvecklar resistens mot cytostatika. Denna studie ställer en avgörande fråga: när kraftfulla läkemedel är utformade för att tvinga cancerceller in i ett dödligt delningsfel, hur hittar cellerna ändå sätt att överleva, och kan den flyktvägen i sig göras till en svaghet? Genom att följa äggstockscellernas utveckling över veckor till månader av behandling avslöjar forskarna ett centralt styrsystem i cellerna som avgör om de fortsätter dela sig eller drar sig tillbaka för att reparera skador — och visar ett nytt sätt att angripa tumörer som redan lärt sig motstå standardläkemedel. 
Två vanliga läkemedel, en gemensam flyktväg
Teamet fokuserade på höggradigt seröst äggstockscancer, den mest aggressiva och vanliga formen av sjukdomen, som nästan alltid bär mutationer i TP53‑genen, det så kallade ”genomets väktare”. Eftersom TP53 är defekt är dessa tumörer ovanligt beroende av senare kontrollstationer i cellcykeln. Två läkemedel som ofta används eller testas i detta sammanhang utnyttjar denna svaghet: adavosertib, en experimentell WEE1‑hämmare som tvingar skadade celler för tidigt in i delning, och paklitaxel, ett standardkemoterapeutikum som låser fast det inre skelettet som krävs för att kromosomer ska separera. I teorin bör båda driva cancerceller in i ”mitotisk katastrof” — ett dödligt delningsmisslyckande. Ändå anpassar sig tumörer ofta i kliniken och i laboratoriet. Forskarna skapade långtidsresistenta cellmodeller genom att successivt höja läkemedelsdoserna över månader, vilket bättre efterliknar vad som händer hos patienter än korta, högdosexperiment.
Hur cancerceller ombyggs för att överleva
Med hjälp av avancerad avbildning och ”Cell Painting” — en teknik som färgar flera cellstrukturer samtidigt — såg forskarna att resistenta celler inte bara såg ut som sina tidigare jag. Många hade flera kärnor, omorganiserade inre skelettstrukturer och bildade tätare kluster samt mindre, mer spridda 3D‑spheroider, kännetecken på partiell formförändring känd som epitel‑mesenkymal transition. Dessa fysiska förändringar antydde att cellerna hade kopplat om hur de rörde sig, delade sig och interagerade. Samtidigt visade detaljerad enskildcells‑RNA‑sekvensering att varje läkemedel och cellinje utvecklade sitt eget mönster av förändrade gener och kromosomförändringar. Trots denna genetiska mångfald framträdde ett konsekvent tema: aktiviteten i en tillväxt- och överlevnadsväg centrerad kring PI3K och AKT ökade i de resistenta modellerna, ofta i sällskap med närliggande signalvägar som MAPK och NF‑κB.
En cellulär brytare mellan ”snabb omväg” och ”långsam reparation”
När forskarna gick djupare fann de att detta PI3K/AKT‑system fungerar som en brytare som växlar cancerceller mellan två överlevnadsstrategier. I ett ”snabb‑omvägsläge” inaktiverar hög PI3K/AKT‑aktivitet bromsproteinet FOXO3 och försvagar cellcykelns kontrollpunkter, vilket tillåter cellerna att fortsätta dela sig och undvika de dödliga effekterna av adavosertib eller paklitaxel. I ett motsatt ”långsam‑reparationsläge” är PI3K/AKT‑aktiviteten lägre, FOXO3 förblir aktiv i kärnan, och cellerna saktar ned sin replikation, aktiverar DNA‑reparationsprogram och pumpar ut läkemedel mer effektivt. Anmärkningsvärt nog utlöste tidig korttidsbehandling en skarp våg av PI3K/AKT‑aktivitet i alla modeller; långsiktig resistens stabiliserade sig sedan i antingen snabb‑omvägs‑ eller långsam‑reparations‑tillstånd beroende på tumorns genetiska bakgrund och tidigare signalering. Detta visar att samma centrala nav kan stödja mycket olika flyktvägar. 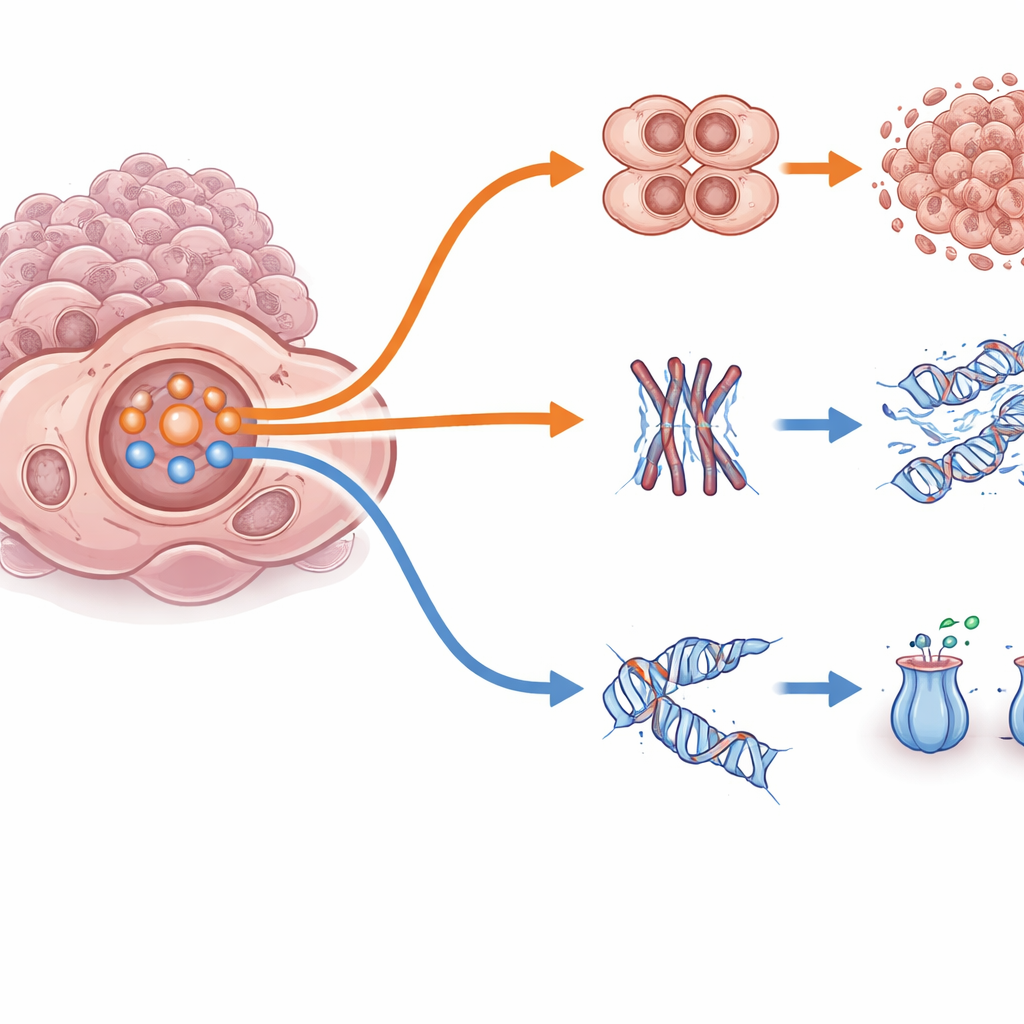
Att göra ett resistenssignal till ett behandlingsmål
En viktig upstream‑aktör i detta nav är ROR1, ett receptorkomplex som normalt är sällsynt i friska vuxna vävnader men förhöjt i flera cancerformer. I många resistenta äggstockscancermodeller steg ROR1‑nivåerna i takt med PI3K/AKT‑aktiviteten. Forskarna visade att upp- eller nedreglering av ROR1 kunde förändra hur lätt cellerna utvecklade resistens mot adavosertib eller paklitaxel, på ett kontextberoende sätt. Viktigast var att de testade zilovertamab‑vedotin, en antikropp‑läkemedelskonjugat som hittar ROR1 och levererar en toxisk last. I både cellinjer och patient‑härledda 3D‑organoider var ROR1‑höga, adavosertib‑resistenta tumörer särskilt känsliga för detta medel, och kombinationen med adavosertib ökade ofta celldöd. Vissa paklitaxel‑resistenta modeller svarade sämre, troligen eftersom de också förstärkt sin förmåga att pumpa ut läkemedel.
Vad detta innebär för framtida behandling av äggstockscancer
Detta arbete omformulerar läkemedelsresistens vid TP53‑muterad äggstockscancer inte som ett slumpmässigt fenomen utan som ett samordnat svar styrt av en central signalbrytare. Genom att identifiera PI3K/AKT–FOXO3‑axeln och ROR1 som nyckelnoder pekar studien på praktiska strategier: kombinera mitos‑inriktade läkemedel som adavosertib och paklitaxel med terapier som blockerar resistensnavet eller utnyttjar ROR1 på resistenta celler. Eftersom ROR1 till stor del saknas i normala vävnader kan sådana kombinationer selektivt angripa återkommande, läkemedelshärdade tumörer samtidigt som normala celler sparas. Även om dessa resultat härrör från laboratoriemodeller och patient‑härledda odlingar snarare än avslutade kliniska prövningar, erbjuder de en tydlig färdplan för att utforma smartare, mer hållbara behandlingar för kvinnor med höggradigt seröst äggstockscancer.
Citering: Raivola, J., Rantanen, F., Dini, A. et al. ROR1-PI3K/AKT signaling drives adaptive resistance to cell cycle blockade in TP53 mutated ovarian cancer. Cell Death Dis 17, 276 (2026). https://doi.org/10.1038/s41419-026-08501-x
Nyckelord: äggstockscancer, läkemedelsresistens, PI3K AKT‑väg, ROR1 antikroppsbehandling, hämare av cellcykeln