Clear Sky Science · sv
Semaphorin 6D driver antitumöriga typ I-interferon-svar för att omprogrammera tumörmikromiljön vid kolorektal cancer
Varför denna forskning är viktig för personer med koloncancer
Kolorektal cancer är en av de ledande orsakerna till cancerrelaterad död globalt, delvis eftersom många tumörer är motståndskraftiga mot dagens behandlingar, inklusive avancerade immunoterapier. Denna studie avslöjar ett naturligt ”bromssystem” inne i kolonstumörer som ofta är avstängt, och visar hur man genom att återaktivera det kan locka tillbaka immunsystemet i kampen. Att förstå denna dolda kontrollomkopplare kan hjälpa läkare att bättre förutsäga prognos och utforma kombinationsbehandlingar som gör immunoterapi effektiv för många fler patienter.
En tyst väktare inne i tumörceller
I centrum för arbetet står en molekyl kallad Semaphorin 6D (SEMA6D), ursprungligen känd för att styra nervväxt och forma det utvecklande hjärtat. Forskarna fann att SEMA6D faktiskt fungerar som en tumörsuppressor i kolorektal cancer: i frisk kolonvävnad finns den närvarande, men i cancerös vävnad är nivåerna markant reducerade. Över flera patientdatamängder och tumörprover kopplades låg SEMA6D till större tumörer, djupare invasion, fler metastaser och signifikant sämre överlevnad. Detta mönster kvarstod även när andra kliniska faktorer beaktades, vilket indikerar att SEMA6D är en oberoende markör för hur aggressiv en kolorektal tumör är.
Hur tumörer stänger av detta skydd
Teamet undrade sedan varför SEMA6D så ofta saknas i tumörer. De upptäckte att genen ofta stängs av genom en kemisk modifiering kallad promotor-hypermetylering—extra kemiska märken som läggs till genens kontrollregion och fungerar som molekylärt tejp över en ljusbrytare. Med detaljerad DNA-kartläggning visade de att nyckelavsnitt i SEMA6D:s kontrollregion är kraftigt metylerade i cancerceller men inte i normala kolonceller. När de behandlade cancerceller med ett avmetylerande läkemedel som används vid blodcancer, togs metylmärkena bort och SEMA6D-produktionen återställdes. De lägsta SEMA6D-nivåerna återfanns i koloncancersubtyper redan kända för omfattande DNA-metylering, hög genetisk instabilitet och stark spridningstendens, vilket stärker kopplingen mellan denna tystnadsmekanism och aggressiv sjukdom.
Från tillväxthämning till immunstärkning
Att återställa SEMA6D förändrade tumörbeteendet på två nivåer. För det första, på cancercellnivå, saktade tvångsuttryck av SEMA6D ner cellernas tillväxt, minskade deras förmåga att migrera och invadera, och reverserade egenskaper hos epitel–mesenkymal transition, ett program som hjälper tumörer att sprida sig. I odlingsskålar och tredimensionella organoider odlade från patienttumörer bildade celler med mer SEMA6D färre och mindre kolonier och visade fler tecken på programmerad celldöd. Hos möss växte tumörer som konstruerats för att överproducera SEMA6D långsammare och gav färre lung- och levermetastaser, medan nedreglering av SEMA6D gav motsatt effekt. För det andra, på immunnivå, innehöll SEMA6D-rika tumörer hos immunokompetenta möss många fler CD4- och CD8-T-celler—de huvudsakliga anfallsstyrkorna i den adaptiva immuniteten—medan SEMA6D-fattiga tumörer var relativt tomma på dessa försvarare. När forskarna eliminerade T-celler försvann till stor del SEMA6D:s tillväxthämmande effekt, vilket visar att mycket av dess kraft kommer från att mobilisera immunsystemet.
Avkodning av den interna larmsignalen
Genom att gå djupare kartlade studien de molekylära stegen som kopplar SEMA6D till immunaktivering. På tumörcellens yta signalerar SEMA6D via en partnerreceptor kallad Plexin A4. Inne i cellen interagerar detta par fysiskt med ett protein som heter IRF9, en nyckelkomponent i maskineriet som svarar på typ I-interferoner—samma antivirala larmsignaler som celler använder för att bekämpa infektioner. När SEMA6D är närvarande och Plexin A4 är intakt aktiveras IRF9 och dess partners, slår på uppsättningar av interferonstimulerade gener och hjälper tumörcellen att sända ut signaler som lockar och rustar T-celler. Avlägsnande av SEMA6D eller Plexin A4 bryter denna kedja och dämpar larmet; återställande av IRF9 kan delvis rädda effekten. Hos möss hade tumörer med aktiv SEMA6D–Plexin A4–IRF9-signalering fler infiltrerande T-celler och lägre nivåer av proliferationsmarkören Ki-67, i linje med starkare immunologiskt tryck på cancern.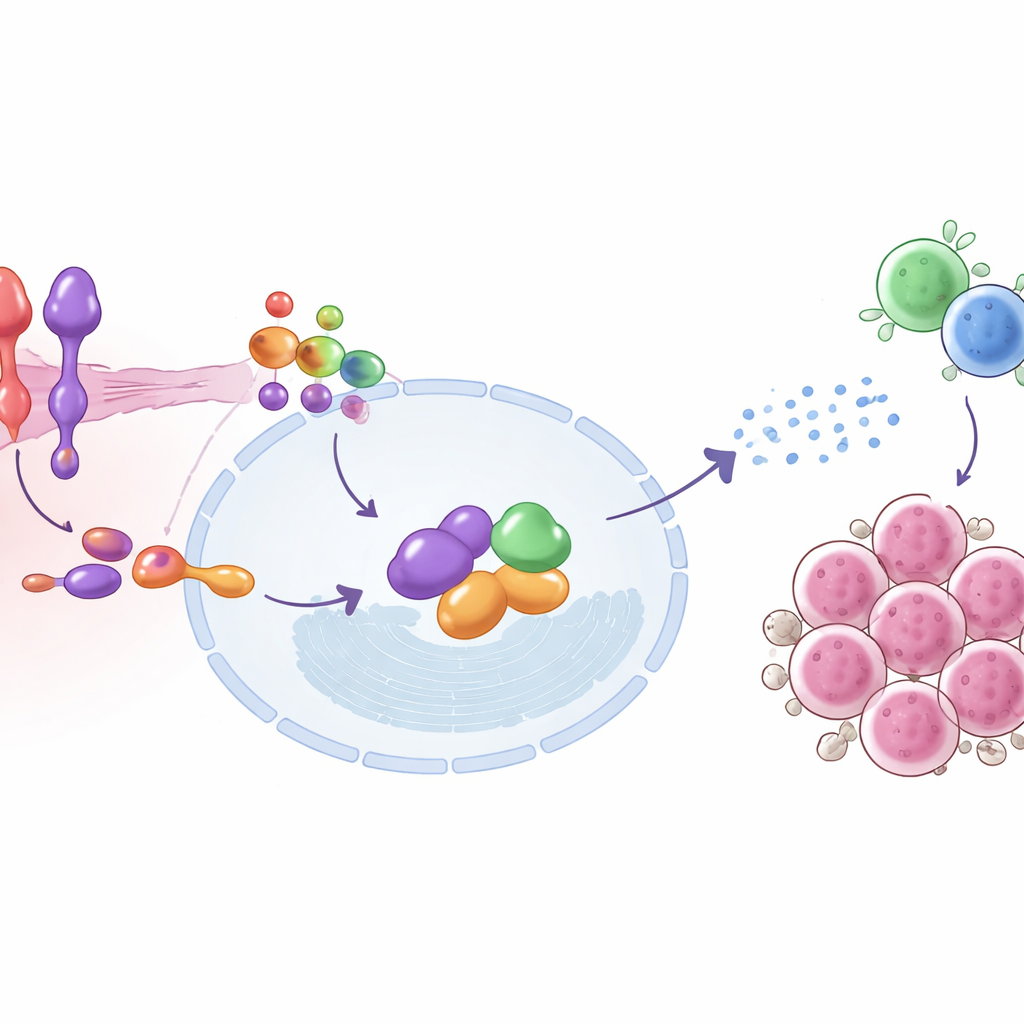
Återväckande av immunitet med kombinationsterapi
Eftersom SEMA6D tystas av metylering testade författarna om ett hypometylerande läkemedel kunde reaktivera det i levande tumörer och därigenom förbättra svaren på immuncheckpoint-blockad. I möss med kolontumörer som behandlades med decitabin följt av en anti–PD-1-antikropp växte tumörerna mycket långsammare än vid någon av behandlingarna ensam. Kombinationen ökade SEMA6D-nivåerna, stärkte interferonvägaktiviteter, minskade cellproliferation och ökade T-cellssinfiltrationen. Dessa resultat tyder på att epigenetiska läkemedel, genom att avlägsna metylerings"lås" från immunkritiska gener som SEMA6D, kan göra immunologiskt ”kalla” tumörer mer ”varma” och därmed mer sårbara för checkpoint-hämmare.
Vad detta betyder för framtida vård
För en lekman är slutsatsen att vissa koloncancerformer gömmer sig för immunsystemet genom att kemiskt stänga av en inbyggd farosignal. Detta arbete identifierar SEMA6D både som den signalen och som ett lovande terapeutiskt handtag. Att mäta SEMA6D och dess metyleringsstatus kan hjälpa till att klassificera tumörer, förutsäga utfall och vägleda behandlingsval. Lika viktigt erbjuder studien en tydlig biologisk motivering för att kombinera DNA-avmetylerande medel med immunoterapi för att återväcka immunövervakning hos patienter vars tumörer i dag inte svarar. Även om kliniska prövningar fortfarande krävs kan denna strategi en dag utvidga fördelarna med immunoterapi till en mycket större grupp personer med kolorektal cancer.
Citering: Shi, W., Zhang, F., Sun, WQ. et al. Semaphorin 6D drives anti-tumor type I interferon responses to reprogram the tumor microenvironment in colorectal cancer. Cell Death Dis 17, 256 (2026). https://doi.org/10.1038/s41419-026-08478-7
Nyckelord: kolorektal cancer, tumörmikromiljö, epigenetisk terapi, typ I-interferon, tumörimmunologi