Clear Sky Science · sv
Obalans i förhållandet α-ketoglutarat/succinat försämrar thymine DNA-glykosylas funktion och bas-excisionsreparation och ökar känsligheten för bukspottkörtelcancer
När vardaglig metabolism möter dold DNA-skada
Bukspottkörtelcancer är en av de dödligaste cancerformerna delvis därför att den ofta upptäcks för sent. Denna studie utforskar en subtil men kraftfull idé: hur långvariga problem som fetma, högt blodsocker och fettrik kost kan tyst omforma kemin inne i bukspottkörtelceller, försvaga deras DNA-reparationssystem och göra det lättare för cancer att få fäste. Genom att följa spåren av små molekyler inne i cellerna visar forskarna hur en rubbad metabol balans kan få DNA att gå från väl underhållet till farligt sårbart.
Hur dietdriven metabolism förbereder bukspottkörteln
För att förstå hur ett ohälsosamt metaboliskt tillstånd påverkar bukspottkörteln använde forskarna möss som genetiskt var benägna att utveckla pankreaslesioner och gav dem antingen en lågfetts- eller högfettsdiet. Högfettkosten framkallade snabbt viktökning, förhöjt blodsocker och kolesterol samt förändringar i gener kopplade till tillväxt och inflammation. På vävnadsnivå visade bukspottkörtlarna hos högfettsfodrade möss tidigare och mer uttalade förstadier till cancer jämfört med kontroller. Metabolitprofilering avslöjade att vissa fetter ändrade i mängd och att molekyler involverade i en- kolmetabolism och metylgruppsdonation — kemiska processer som påverkar hur DNA märks och läses — var påtagligt förändrade, särskilt den viktiga metylgruppen-donatorn S-adenosylmetionin.
Små molekyler som styr DNA-märkning
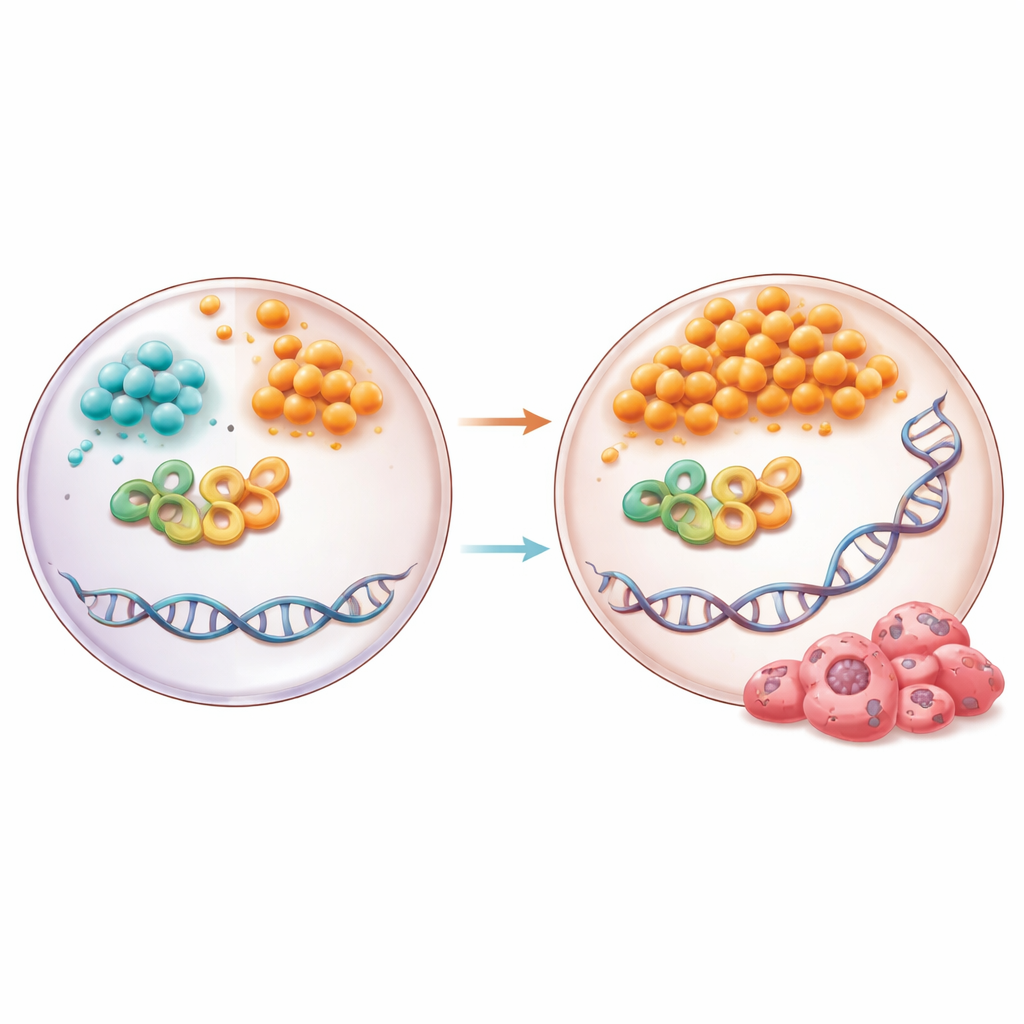
Inne i celler sätts och tas de kemiska ”taggarna” på DNA bort av enzymer som är beroende av specifika metaboliter som bränsle eller medhjälpare. I detta arbete framträdde balansen mellan två molekyler ur cellens energicykel, alfa-ketoglutarat och succinat, som avgörande. I miljön med högfettdiet och i humana pankreasgångsceller exponerade för högt glukos och ett vanligt dietfett sjönk nivåerna av alfa-ketoglutarat medan succinat steg, vilket snedvridde deras förhållande. Denna förändring störde ett DNA-demetyleringspartnerskap mellan enzymet TET1 och reparationsproteinet TDG. Istället för att smidigt växla DNA-markeringar uppnådde pankreasepitelceller en ansamling av intermediära cytosinmodifieringar och kemiska märken på DNA kända som abasiska platser — luckor där en bas saknas.
När reparationshjälpare blir överaktiva
Gruppen grävde djupare i hur succinat kan påverka TDG självt. Genom datorbaserade simuleringar, biofysiska experiment och enzymtester fann de att succinat binder direkt till en kritisk plats på TDG, samma region som används av alfa-ketoglutarat. Till skillnad från alfa-ketoglutarat tryckte succinat dock TDG in i ett hyperaktivt tillstånd. Denna överaktivitet ledde till ett överskott av DNA-ställen där baser klippts ut men ännu inte reparerats korrekt. I pankreasceller odlade under dysmetabola förhållanden, eller behandlade med cellpermeabelt succinat, ökade både TDG-aktivitet och ansamling av abasiska platser, och detta var beroende av den specifika TDG-plats som känner av dessa metaboliter.
En bruten mållinje för DNA-reparation
Normalt, efter att TDG och närbesläktade enzymer skapar en tom plats i DNA, träder en reparationsväg kallad bas-excisionsreparation in för att fylla och sammanfoga luckan. Två DNA-ligaset, LIG1 och LIG3, utför det avgörande sista hopfogandet. Både hos högfettsfodrade möss och i metabolt stressade humana pankreasceller föll nivåerna av LIG1 och LIG3 kraftigt. Studien visar att deras genpromotorer blir mer metylerade i denna miljö, sannolikt därför att metylgruppsdoneringen är överaktiv medan demetyleringen sviktar. Som en följd dämpas ligasernas uttryck när de behövs som mest, vilket får reparationen att stanna av och abasiska platser att ansamlas. När forskarna hämmade ligaser direkt ökade antalet abasiska platser, vilket understryker hur viktiga dessa enzymer är för att förhindra att DNA-skador byggs upp.
Kan skadan återställas?
Anmärkningsvärt nog återställde tillskott av alfa-ketoglutarat i metabolt stressade pankreasceller delvis detta skadliga program. Extra alfa-ketoglutarat minskade metylering på LIG1- och LIG3-promotorerna, återupplivade deras uttryck och sänkte antalet abasiska platser. Tillsammans målar fynden upp en bild av en metabol-epigenetisk axel: kronisk dysmetabolism höjer succinat och metylgivare, stör TET1–TDG-samarbetet, hyperaktiverar TDG och tystar kritiska reparationsligaser. Resultatet blir instabilt DNA i snabbt delande pankreasceller, vilket kan bana väg för förstadier till cancer. För icke-specialister antyder arbetet att metabolismens kemi inte bara påverkar vikt och blodsocker; den kan tyst omforma hur vårt DNA underhålls och potentiellt öppna nya möjligheter för tidiga biomarkörer och metabo-riktade strategier för att minska risken för bukspottkörtelcancer.
Citering: Malatesta, S., Vigiano Benedetti, V., Salviati, E. et al. α-ketoglutarate/succinate ratio imbalance impairs thymine DNA glycosylase function and base excision repair process increasing susceptibility to pancreatic cancer. Cell Death Dis 17, 242 (2026). https://doi.org/10.1038/s41419-026-08475-w
Nyckelord: bukspottkörtelcancer, metabolism, DNA-reparation, epigenetik, succinat