Clear Sky Science · sv
BCL-xL som terapeutiskt mål vid cetuximab-refraktär kolorektal cancer
Varför detta är viktigt för personer med tjock- och ändtarmscancer
Kolorektal cancer (tjock- och ändtarmscancer) är en av de vanligaste cancerformerna i världen, och många patienter med avancerad sjukdom får en målinriktad antikroppsbehandling som heter cetuximab. Denna behandling kan initialt krympa tumörer, men hos de flesta patienter hittar cancern sätt att undkomma inom månader, vilket lämnar få bra behandlingsalternativ. Denna studie ställer en brännande fråga: när kolonstumörer slutar svara på cetuximab, finns det en annan svag punkt som nya läkemedel kan utnyttja för att få cancercellerna att dö?
När ett målinriktat läkemedel slutar fungera
Cetuximab verkar genom att blockera en ytreceptor på cancerceller kallad epidermal tillväxtfaktorreceptor (EGFR), som bidrar till deras tillväxt. Forskarna skapade en laboratoriemodell för resistens genom att utsätta en känslig kolorektal cancercellinje (LIM1215) för gradvis ökande doser cetuximab under sex månader. Två oberoende framväxande resistenta cellpopulationer uppstod som fortsatte växa även vid höga läkemedelsnivåer, men såg lika friska och snabbväxande ut som ursprungscellerna när läkemedlet togs bort. Viktigt var att de resistenta cellerna fortfarande bar läkemedlets mål på ytan och cetuximab kunde fortfarande binda till det, vilket tyder på att cancern inte enkelt hade ”gömt” eller förändrat receptorn.
Resistenta celler omdirigerar sina tillväxtsignaler
För att förstå hur cellerna kringgick cetuximab undersökte forskarna nyckelvägar för tillväxt inuti cellen. I föräldracellerna dämpade cetuximab normalt MAPK-vägen, en viktig drivkraft för celldelning. I de resistenta cellerna förblev MAPK-aktiviteten hög även när EGFR blockerades, vilket visar att tillväxtsignalen hade kopplats loss från den ursprungliga receptorn. Sekvensering av cellernas RNA avslöjade nya aktiverande mutationer i en annan RAS-gen, HRAS, i subpopulationer av de resistenta cellerna, men inte i andra vanliga kandidater som KRAS, NRAS eller BRAF. Försök att stänga ner denna omdirigerade signalering med en MEK-hämmare (som verkar nedströms om RAS) minskade tillväxten endast måttligt. Detta betonade att det, istället för att jaga varje ny mutation, kan vara mer effektivt att angripa cellens liv-eller-död-mekanismer som delas av olika resistenta kloner. 
Att slå mot cancer i dess livsuppehållande system
Forskarna vände fokus mot apoptos, det inbyggda cellsjälvmordsprogram som cancer ofta undertrycker. Genuttrycksanalys visade att apoptosrelaterade vägar var förändrade i de resistenta cellerna. Särskilt var det anti-dödsproteinet BCL-xL högre i en resistent population och måttligt ökat i den andra, samtidigt som ett annat överlevnadsprotein, MCL-1, också fanns närvarande. Teamet testade småmolekylära läkemedel kallade BH3-mimetika som är utformade för att blockera dessa överlevnadsproteiner och frigöra dödsmaskineriet. I tvådimensionella cellkulturer var alla tre cellinjerna—föräldra- och resistenta—känsliga för läkemedel som blockerar BCL-xL eller MCL-1, men anmärkningsvärt nog dödade lägre doser de cetuximab-resistenta cellerna effektivare. Tillsats av en låg dos av proteasomhämmaren bortezomib, som hjälper till att ackumulera pro-dödsignaler, ökade dödligheten ytterligare, särskilt i kombination med MCL-1-hämmaren.
Från plana skålar till 3D-mini-tumörer och patientvävnader
Då platta cellskikt inte fullt ut kan efterlikna tumörer i kroppen odlade teamet cellerna som tredimensionella spheroider inbäddade i en gel, vilket bättre speglar arkitekturen och läkemedelspenetrationsutmaningarna i verkliga tumörer. Återigen minskade blockad av BCL-xL eller MCL-1 spheroidernas viabilitet, och kombinationen av dessa läkemedel med bortezomib orsakade dramatiska minskningar i metabolisk aktivitet och tydliga tecken på celldöd. För att testa om denna sårbarhet finns i mer realistiskt mänskligt tumörmaterial använde de tunna skivor av cetuximab-resistenta kolorektala tumörer odlade i möss från patienttumörer (patientderiverade xenograft-modeller). Dessa modeller var alla KRAS vildtyp likt de ursprungliga LIM1215-cellerna men bar olika ytterligare mutationer, inklusive i BRAF och TP53, vilket speglar den genetiska mångfald som ses i kliniken. 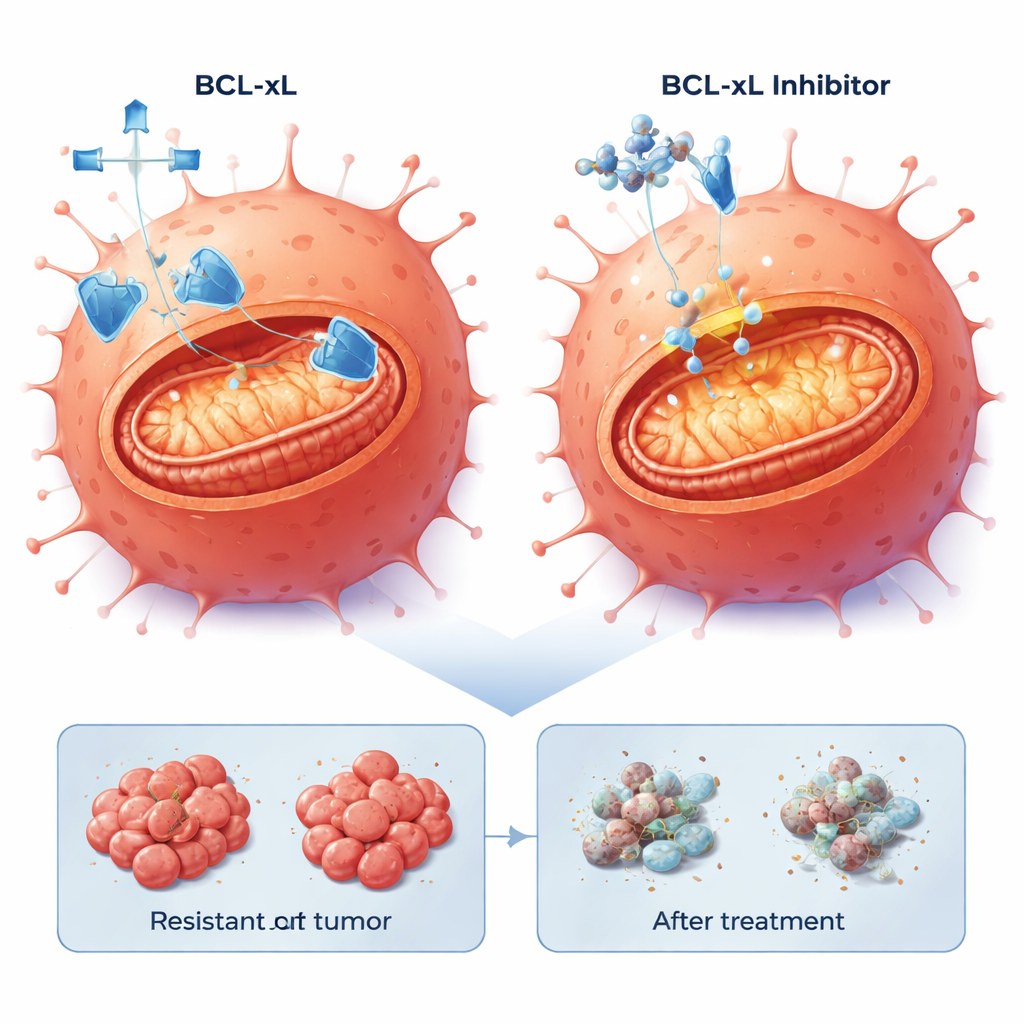
Att rikta in sig på BCL-xL fungerar över olika resistenta tumörer
I dessa patientderiverade tumörskivor utlöste kombinationen av en BCL-xL-hämmare med lågdoserat bortezomib konsekvent kraftig celldöd i 20–40 % av tumörcellerna över fyra olika modeller, inklusive de med aggressiva BRAF-mutationer. Däremot gav blockad av MCL-1 med bortezomib starka effekter endast i en delmängd av tumörerna. Viktigt är att de resistenta cellernas förmåga att genomgå apoptos bevarades: när BCL-xL-säkerhetsnätet togs bort kunde det interna dödsprogrammet fortfarande aktiveras, oberoende av vilken genetisk väg tumören tagit för att undkomma cetuximab.
Vad detta betyder för patienter
För personer vars kolorektala cancer slutar svara på cetuximab erbjuder denna studie försiktig optimism. Den tyder på att även efter att tumörer blivit resistenta mot EGFR-riktad terapi är många cancerceller fortfarande redo att dö om ett nyckelöverlevnadsprotein, BCL-xL, blockeras. Även om BCL-xL-hämmare kan ha biverkningar, särskilt på blodplättar, pekar arbetet mot kombinations- och dosoptimeringsstrategier som kan begränsa toxicitet samtidigt som man utnyttjar en gemensam Achilleshäl hos svårbehandlade tumörer. I framtiden kan läkemedel som oskadliggör BCL-xL utgöra ryggraden i nya andrahandsbehandlingar för cetuximab-refraktär kolorektal cancer, med mindre beroende av tumörens skiftande mutationslandskap.
Citering: Asmanidou, S., Thiel, J., Ekstrom, T.L. et al. BCL-xL as a therapeutic target in cetuximab-refractory colorectal cancer. Cell Death Dis 17, 187 (2026). https://doi.org/10.1038/s41419-026-08434-5
Nyckelord: kolorektal cancer, läkemedelsresistens, cetuximab, BCL-xL-hämning, apoptos