Clear Sky Science · sv
Öka KLF15-aktivitet i kardiomyocyter: en ny metod för att förhindra patologisk omprogrammering och fibros via nukleasdefekt dCas9VPR
Omprogrammera det sviktande hjärtat
Hjärtsvikt drabbar miljontals människor och utvecklas ofta gradvis efter år av högt blodtryck eller klaffsjukdom. Vid dessa tillstånd blir hjärtmuskelceller inte bara större utan aktiverar också ett "fetal" genetiskt program och hjärtat fylls med ärrvävnad. Denna studie undersöker ett nytt sätt att styra hjärtats egna genreglerande maskineri tillbaka mot ett friskare tillstånd — utan att klippa i DNA — genom att varsamt öka en skyddande regulator kallad KLF15 i hjärtmuskelceller.
När hjärtceller förlorar sin identitet
I ett friskt vuxet hjärta förbränner kardiomyocyter — hjärtmuskelceller — fett effektivt för energi och upprätthåller ett stabilt mönster av genaktivitet. Genom single-cell RNA-sekvensering i möss utsatta för kronisk trycköverbelastning kartlade forskarna hur enskilda kardiomyocyter förändras när hjärtat går från normal funktion till förstorning och slutligen svikt. De fann att en transkriptionsfaktor kallad KLF15, som normalt håller metabolism och tillväxt i balans, visade den starkaste aktivitetsförändringen i sjuka celler. När stressen ökade sjönk KLF15-nivåerna och dess förmåga att dämpa fetala och stressrelaterade gener försvagades. Liknande minskningar av KLF15 observerades i mänskliga hjärtan från patienter med dilaterad och hypertrofisk kardiomyopati, vilket tyder på att denna störning är bevarad mellan arter.
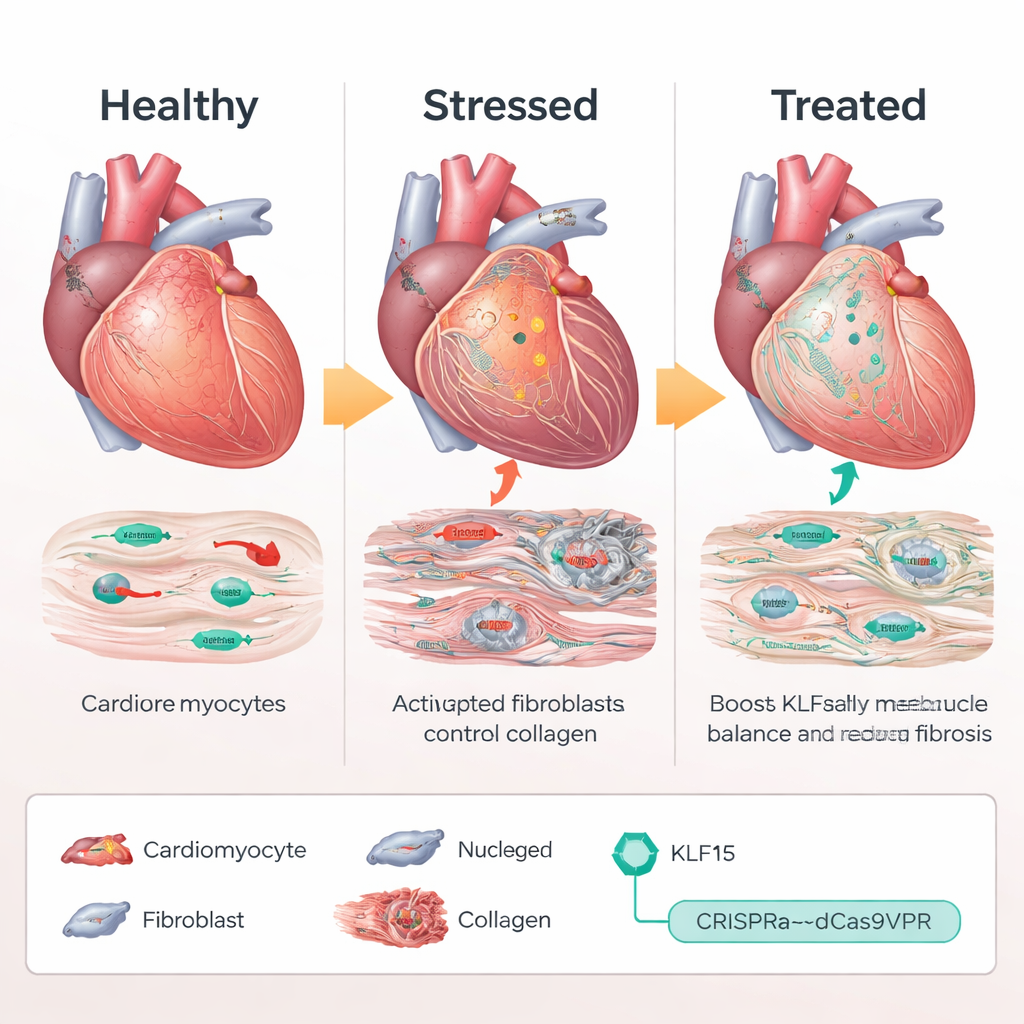
Använda CRISPR som en volymknapp, inte som sax
I stället för att tillföra en extra kopia av KLF15-genen eller skära i DNA använde teamet ett CRISPR-baserat "aktiverings"-system, kallat dCas9VPR, som binder nära den naturliga Klf15-genen och förstärker dess egen uttrycksnivå. I möss konstruerade för att uttrycka denna CRISPR-aktivator endast i kardiomyocyter levererade forskarna guide-RNA med ett adeno-associerat virus (AAV9) för att rikta in sig mot Klf15-promotorn. Under kronisk trycköverbelastning bibehöll mössen som fick Klf15-aktiverande guider nära-normala Klf15-nivåer. Deras hjärtmuskelceller förblev mindre, pumpfunktionen minskade mindre och överlevnaden förbättrades jämfört med kontrollgrupper. På molekylär nivå dämpades stress- och fetala gener, medan genuttryck för metabolism och kalciumhantering återhämtade sig, vilket tyder på att det skadliga transkriptionsprogrammet till stor del återställdes.
Att dämpa ärrbildning via cell-till-cell-signalering
Hjärtsvikt drivs inte bara av sjuka muskelceller utan också av fibroblaster, stödjeceller som producerar kollagen och bildar stel ärrvävnad. Single-cell-analyser och vävnadsavbildning visade att återställande av Klf15 i kardiomyocyter minskade fibroblastaktivering och den totala fibrosen, trots att genterapin aldrig riktades direkt mot fibroblaster. Teamet spårade denna effekt till ett sekretoriskt protein kallat AZGP1. När Klf15 ökades i kardiomyocyter ökade produktionen och frisättningen av AZGP1. I både mus-hjärtan och humana stamcellsderiverade hjärtvävnader dämpade högre AZGP1 TGF-β / SMAD-vägen i fibroblaster — en nyckeldrivare av ärrbildning — och sänkte nivåerna av markörer som α-SMA och POSTN. Viktigt är att överuttryck av AZGP1 enbart i kardiomyocyter inte omprogrammerade muskelcellerna, vilket visar att KLF15 främst skyddar kardiomyocyter direkt och använder AZGP1 som ett budbärarmolekyl för att hålla fibroblasterna i schack.
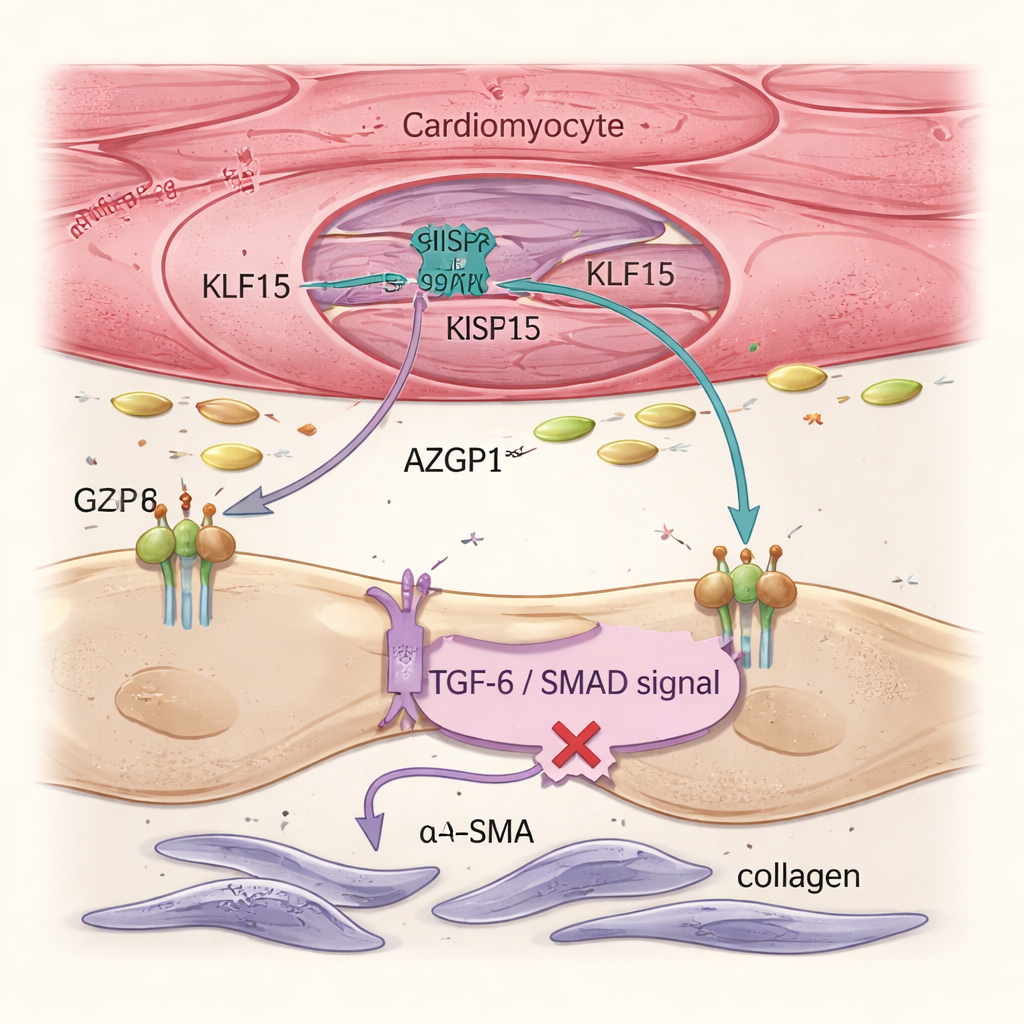
Human vävnadsmodell bekräftar den skyddande kretsen
För att testa om dessa mekanismer gäller i mänskliga celler använde forskarna inducerade pluripotenta stamcells-deriverade kardiomyocyter odlade i tredimensionella konstruerade hjärtvävnader. När dessa utsattes för mekanisk belastning som efterliknar högt blodtryck förlorade vävnaderna KLF15, aktiverade stress- och fetala gener, stelnade och deras kontraktioner försvagades — vilket återgav sjukdomsdragen. CRISPRa-driven återställning av KLF15 förhindrade denna försämring, bevarade kraftgenereringen och skiftade genuttrycket tillbaka mot mogen metabolism och struktur. Detaljerade experiment visade att TGF-β1, en välkänd profibrotisk signal, reducerar KLF15 i mänskliga kardiomyocyter via SMAD2/3-vägen, vilket hjälper förklara hur kronisk stress leder till maladaptiv ombyggnad. Slutligen konstruerade teamet ett kompakt, "mini"-CRISPRa-system baserat på en mindre Cas9-variant som rymdes i ett enda AAV9-vektor och drivs av en kardiomyocyt-specifik promotor. I precisionsskurna skivor av sviktande mänsklig hjärtvävnad ökade denna vektor framgångsrikt KLF15-nivåerna och förbättrade kontraktil prestanda över dagar i odling.
En ritning för mildare genterapi
För en icke-specialist är kärnbudskapet att detta arbete visar hur noggrant uppreglering av en enda skyddande regulator inne i hjärtmuskelceller både kan stabilisera deras identitet och sända signaler som begränsar ärrbildning. Genom att använda en CRISPR-baserad aktivator som inte skär DNA finjusterar metoden hjärtats egna genuttryck istället för att införa en artificiell gen. Studien definierar en TGF-β → KLF15 → AZGP1-väg som kopplar mekanisk stress till skadlig ombyggnad och visar, i möss, mänskliga cellmodeller och mänskliga hjärtvävnadsskivor, att återställande av KLF15 kan bryta denna kedjereaktion. Även om arbetet fortfarande är i preklinisk fas erbjuder det kompakta, kardiomyocyt-riktade CRISPRa-systemet här ett potentiellt vägledande koncept för behandling av vanliga, icke-genetiska former av hjärtsvikt genom att omprogrammera genaktivitet snarare än att skriva om genomet.
Citering: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Nyckelord: hjärtsvikt, KLF15, CRISPR-aktivering, kardial fibros, AZGP1